 一文詳解鋰離子電池技術
一文詳解鋰離子電池技術

文章來源:老千和他的朋友們???
原文作者:孫千???
本文主要介紹鋰離子電池技術。??????
鋰離子電池的起源
化學元素鋰于1817年由Johan August Arfwedson通過分析礦物鋰長石(LiAlSi?O??)發現,并于1818年由J?ns Jakob Berzelius首次報道。1821年,William Thomas Brande通過電解鋰氧化物(Li?O)成功分離出鋰。
Lewis和Keyes于1913年通過鋰/汞金屬間化合物開始研究鋰金屬的電化學性質。在丙胺溶液中的LiI中,測量了鋰、鋰/汞金屬間化合物和甘汞(Hg?Cl?)電極之間的相對電位。鋰金屬的電位為3.3044V(相對于甘汞),被描述為"迄今為止測得的最高電極電位"。這一說法至今仍然成立,并為所有現代基于鋰金屬或鋰衍生物的電池研究奠定了基礎。
對高能鋰基電池的追求始于20世紀50年代。1958年,由于使用鋰時必須避免水和空氣(因其具有反應性),William S. Harris研究了鹽在各種非水電解質中的溶解度和導電性——包括環狀酯(碳酸乙烯酯、碳酸丙烯酯、γ-丁內酯和γ-戊內酯)、熔融鹽和溶解在碳酸丙烯酯中的無機鋰鹽(LiClO?)。他觀察到形成了一種鈍化層,能夠防止鋰與電解質之間的直接化學反應,同時仍允許離子傳輸,這導致了對鋰離子電池穩定性的研究。這些研究也增加了對一次性鋰離子電池商業化的興趣。
自20世紀60年代末以來,非水性3V鋰離子一次電池已經在市場上出現,使用不同的正極,包括鋰-二氧化硫電池、鋰-一氟化碳電池、鋰-二氧化錳電池和鋰-鹵素電池。同時,對使用不同材料的鋰離子一次電池的理解進步催生了可充電(二次)鋰離子電池。一些綜述論文中也可以看到最終導致鋰離子電池誕生的關鍵發現和技術成就的廣泛描述。
鋰離子電池的歷史??
室溫下可充電鋰離子電池的廣泛研究始于20世紀70年代初,在發現了能量儲存中的嵌入反應之后。在化學中,嵌入是離子或分子可逆地插入晶格結構而不顯著改變晶格結構的過程,除了輕微的膨脹或收縮。最初,研究人員通過將離子或有機分子嵌入到主體材料的結構中,修改載流子密度,從而創造了超導體。
M.Stanley Whittingham展示了第一個可充電鋰離子電池,使用層狀TiS?作為正極,鋰金屬作為負極,以及二氧戊環中的LiClO?作為電解質。在展示基于TiS?的鋰離子電池之后,各個研究小組研究了不同的金屬硫族化物作為鋰離子電池的電極材料。
事實上,大多數鋰離子電池在兩個電極上都利用嵌入反應進行能量儲存,表明基礎技術在近40年內沒有改變。此外,鋰-鋁(Al)合金被用作負極,它是通過將鋰片放在鋁片頂部形成的。在電池構建的最后階段,在電池密封前,添加電解質,使鋰和鋁片之間的反應形成鋰-鋁合金。
1980年,John Goodenough首次提出利用層狀鈷酸鋰(LiCoO?)作為高能量和高電壓正極材料。1983年,Goodenough還確定了錳尖晶石(LiMn?O?)作為低成本正極材料。然而,缺乏安全的負極材料限制了層狀氧化物正極在鋰離子電池中的應用。1987年,Yohsino等人申請了專利并建造了使用碳質負極和LiCoO?正極的原型電池。碳負極和LiCoO?正極在空氣中都很穩定,這對工程和制造非常有利。
索尼在20世紀90年代初成功將第一款基于碳負極(石油焦)和LiCoO?正極的可充電鋰離子電池商業化。設計的電池展示了>3.6V的開路電壓和約150 Wh/kg的能量密度。從那時起,很多研究一直在進行并且仍在進行中,以改善電極材料的性能。
鋰離子電池的基本結構
雖然探索了各種類型的電極材料、電解質和隔膜,但鋰離子電池的基本設計仍與M. Stanley Whittingham在1976年提出的相同。通常,鋰離子電池主要由正極和負極材料、集流體、電解質和隔膜組成。
為了將正極和負極材料(通常稱為活性材料)涂覆在集流體上,通常先制備漿料。漿料是通過將液體溶劑、聚合物粘合劑、導電添加劑和活性材料混排在一起制備的。正極和負極通過隔膜相互隔離。隔膜在電極之間提供電絕緣,同時允許鋰離子在電解質相中通過隔膜傳輸。
原則上,電解質應該具有離子導電性和電子絕緣性;然而,電解質的實際行為要復雜得多。嵌入/脫嵌是可充電電池中最經典的電荷儲存機制。
在石墨負極和LiCoO?正極的情況下,如圖1所示,在充電過程中,鋰離子從層狀LiCoO?主體中脫嵌,穿過電解質并嵌入石墨層之間。這一過程在放電過程中逆轉。充放電過程中的電化學反應可以簡要描述如下:
正極:LiCoO? Li???CoO? + xLi? + xe?
負極:6C + xLi? + xe? Li?C?
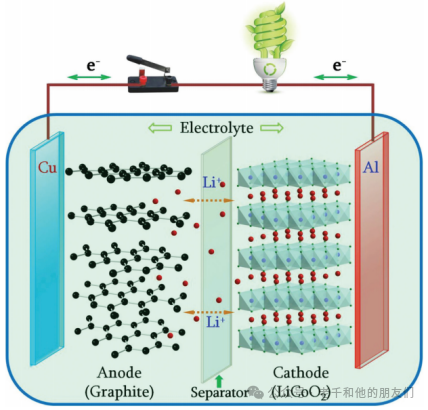
圖1鋰離子電池結構示意圖
鋰離子電池的尺寸取決于應用和功率規格。鋰離子電池有不同形式,如紐扣電池、圓柱形電池、棱柱形電池和袋裝電池(圖2)。紐扣電池主要用于不可充電的便攜設備,包括醫療植入物、手表、助聽器、汽車鑰匙和內存備份。圓柱形電池的典型應用是電動工具、醫療儀器、筆記本電腦和電動自行車。棱柱形電池主要用于手機、平板電腦和薄型筆記本電腦。袋裝電池為電池設計提供了簡單、靈活和輕量級的解決方案。
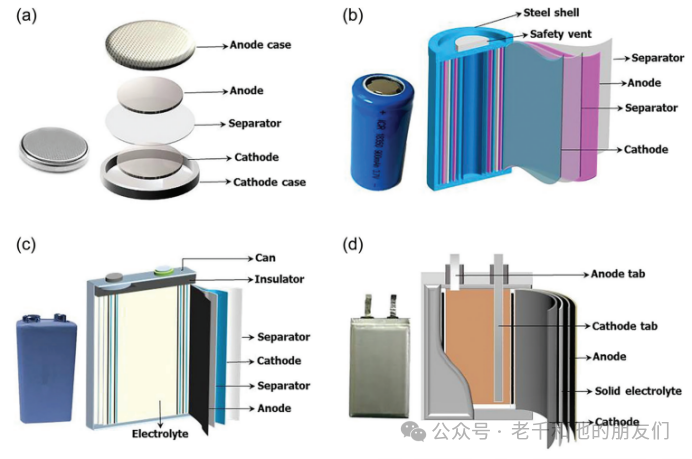
圖2 典型可充電電池配置的示意圖:(a)紐扣型,(b)圓柱型,(c)棱柱型和(d)軟包型。
超越鋰離子電池
盡管新型電池技術可能無法在市場份額上直接與鋰離子電池競爭,因為它們在性能、成本和尺寸方面具有獨特特性,但一些有前途的電池系統正在為未來應用而開發。
為了開發高能量密度的電池,鋰硫(Li-S)和鋰空氣(Li-O?)電池因其最高的理論能量密度而受到廣泛的研究關注。由于非鋰金屬比鋰更豐富,非鋰金屬離子電池也已經出現并在能源存儲應用中顯示出巨大潛力。例如,鈉離子電池(SIBs)是最有前途的"超越鋰"能源存儲技術之一,因為鋰離子電池的大部分專業知識可以直接用于鈉離子電池的開發。同樣,鎂離子電池(MIBs)通過避免枝晶形成而具有增強安全性的優勢,并且在顯著降低成本的同時能夠達到與鋰離子電池相當的能量密度。此外,鋅離子電池(ZIBs)可以被視為電網能源存儲最有前途的替代方案之一。最后,鋁離子電池(AIBs)通過三電子轉移反應,理論上可以實現比鋰離子電池高三倍的能量密度。由于豐富的鋁資源和低成本,鋁離子電池已成為最有前景的下一代電池系統之一。
鋰離子電池的正極材料
作為鋰離子電池的關鍵組成部分,高能量密度、長循環壽命和高安全性的正極材料備受青睞。與石墨負極相比,正極的相對較低容量已成為提高鋰離子電池能量密度的瓶頸。因此,已經進行了大量研究來探索具有高能量密度的先進正極材料。
此外,鋰離子電池的其他關鍵性能參數,包括倍率性能和循環壽命,也至關重要,這些由正極材料的內在化學特性決定。例如,倍率性能與正極材料的電子和離子導電性高度相關,而循環壽命則強烈受到正極材料成分和充電狀態的影響。鋰離子電池的正極材料可分為幾類,即層狀正極、尖晶石正極、多陰離子正極、無序巖鹽型正極、轉化型正極、硫和氧氣正極。
層狀正極
層狀正極以特定結構為特征,由鋰離子和金屬化合物的交替層組成。層狀結構是鋰離子電池正極材料中最早形式的嵌入化合物。金屬硫族化合物最先被研究作為可能的嵌入式正極。二硫化鈦(TiS?),一種具有鋰嵌入位點的六方密堆結構,因其高能量密度(約130 Wh/kg或280 Wh/L)和延長的循環壽命而被廣泛研究。
圖3顯示了LiTiS?化合物的層狀結構,鋰離子可以在過渡金屬硫化物層之間嵌入。LiTiS?嵌入化合物是嵌入的理想例子,在x從0到1的所有值范圍內顯示鋰的完全溶解度。圖3還顯示了在電流密度為10mA/cm2時LiTiS?的充放電曲線。
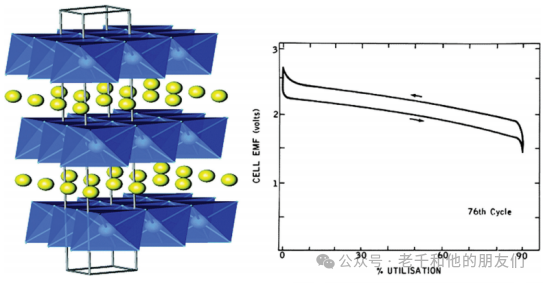
圖3(左)LiTiS?的層狀結構。(右)在電流密度為10mA/cm2時Li/TiS?的充放電曲線。
五氧化二釩(V?O?)和三氧化鉬(MoO?)是最早研究用于可充電鋰電池的兩種層狀過渡金屬氧化物(TMOs)。與層狀二硫族化物具有相同結構的層狀過渡金屬氧化物因其高電壓和高能量密度而被最廣泛研究。
層狀鈷酸鋰(LiCoO?,LCO)是第一種被廣泛研究和商業成功的層狀過渡金屬氧化物正極。LCO由John Goodenough認識并引入,由索尼商業化,與碳質負極結合制造鋰離子電池。
一價鋰離子和三價鈷離子在巖鹽結構的氧化物離子立方密堆陣列的交替(111)平面上有序排列。位于八面體位置的鈷和鋰占據交替層并形成六方對稱性。LCO顯示相對高的理論比容量274mAh/g和高的理論體積容量1,363 mAh/cm3。然而,當從層狀晶格中提取超過一半的鋰離子(x>0.5)時,Li???CoO?會發生一系列不可逆的結構轉變,導致不可逆容量損失。通常,元素摻雜、表面涂層和共同修飾可以有效提高結構穩定性并促進循環穩定性。如今,商業LCO的實際比容量可達185mAh/g,對應于4.5V的充電截止電壓。
LiNiO?(LNO)具有與LiCoO?相同的晶體結構和相似的理論比容量275mAh/g。在Jeff Dahn等人的努力下,相對較高的能量密度和低成本推動了對LNO的研究。純LNO難以合成,大多數報告建議過量的鎳,如Li???Ni???O?,其中Ni2?離子在合成甚至脫鋰過程中傾向于取代Li?位點。實際上,這種鋰/鎳混排是高鎳層狀氧化物中一個臭名昭著的內在問題。此外,LNO還遭受有害的結構轉變,阻塞鋰離子擴散通道,導致循環性能不佳,并且由于高效的平衡氧分壓,在低鋰含量時顯示出較差的熱穩定性。
結合鎳和鈷,研究人員發現LiNi???Co?O?是減少陽離子無序和增加熱穩定性的有效方法。此外,與LNO相比,LiNi???Co?O?相更容易制備,不需要使用氧氣氛圍。
另外,研究LiMnO?(LMO)是因為錳比鈷或鎳便宜得多且毒性較低。純LiMnO?正極在鋰離子電池中的實際使用受到阻礙,這是由于與高自旋Mn3?相關的協同楊-泰勒(John–Teller)效應,導致嚴重的結構退化和快速的容量衰減。結合鎳和錳,一種有趣的材料是LiNi?.?Mn?.?O?,在2.5V-4.3V的電壓范圍內提供穩定的容量160mAh/g。
為了解決LCO和LNO的熱不穩定性,層狀三元過渡金屬氧化物,如LiNi?????Co?Mn?O?(NMC)和LiNi?????Co?Al?O?(NCA),在可逆容量、倍率性能和成本效益方面是最實用的廣泛應用候選者。NMC可以被視為LiNiO?、LiCoO?和LiMnO?的固溶體。圖4展示了三種單獨鋰化氧化物(LiNiO?、LiCoO?和LiMnO?)的相圖,具有鎳、鈷和錳的各種成分。
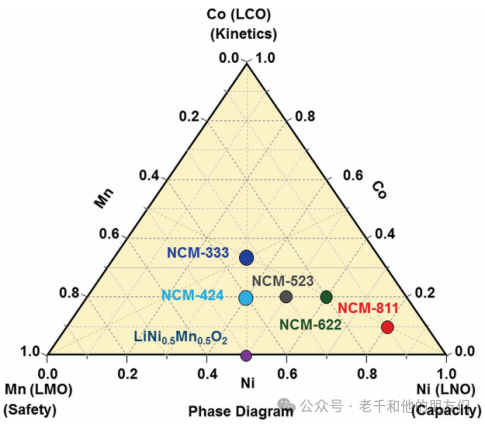
圖
4 由LNO、LCO和LMO生成的三元系統相圖,顯示了一些代表性成分。
在三元金屬氧化物的結構中,每種金屬元素執行其功能并提供協同效應。在NMC中引入錳和在NCA中引入鋁可以保持結構穩定性并降低成本。鎳允許高鋰提取并提供高容量,而鈷防止鎳占據鋰位點,從而保證高可逆容量。
對NMC正極的研究始于不同團隊的低鎳含量。2001年,Ohzuku和Makimura合成并表征了LiNi?.??Mn?.??Co?.??O?(NMC111),而Jeff Dahn及其同事研究了LiNi?Co????Mn?O?(x = 0.25和0.375)。此后,一系列NMC被開發和商業化,包括NMC422、NMC532、NMC622和NMC811。
鎳含量的增加提高了電荷存儲容量。例如,NMC811顯示200mAh/g的可逆容量,而NMC111僅顯示150mAh/g的較低容量。然而,由于高鎳NMC不穩定的材料特性,容量衰減和熱不穩定性加劇。Ni??離子對液體有機電解質和微量水分的不穩定性增加了正極材料的反應活性。
表面涂層和部分元素替代是克服性能退化最常見和有效的方法。表面涂層可以減少電解質和正極材料之間的反應,而元素替代可以影響電子結構以穩定材料并防止在鋰移除過程中結構崩塌。例如,研究人員合成了鈮(Nb)涂層和替代的NMC811以改善電化學性能。鈮涂層穩定了表面,減少了首次循環損失并提高了倍率性能,而鈮替代通過穩定晶格結構改善了長時間循環的容量保持率。
與NMC類似,NCA也可以被視為LiNiO?、LiCoO?和LiAlO?的固溶體。NCA最流行的配方是LiNi?.?Co?.??Al?.??O?,顯示出與NMC811相當的比容量200mAh/g。需要注意的是,NCA中第三種金屬(Al:5%-10%)的含量相對較低,遠低于NMC中的錳(10%-40%)。這是因為使用更高水平的鋁(>10%)會導致嚴重的容量衰減和結構中鋰離子擴散性能下降。
富鋰層狀氧化物,尤其是富鋰錳基正極材料(xLi?MnO?·(1–x)LiMO?,0
圖5 (a)菱方LiMO2相(空間群:R-3m,M = Ni, Co, Mn)和(b)單斜Li2MO3相(空間群:C2/m)的晶體結構
如前文所述,正極含有過渡金屬離子,即Mn、Ni和Co。過渡金屬離子從正極溶解到電解質中并最終沉積在負極上是不可避免的,尤其是在高溫和高電池電壓條件下。過渡金屬離子的溶解是鋰離子電池容量和功率衰減的主要原因之一。
過渡金屬離子的溶解是酸堿反應,如電解質中發現的微量HF所致。這些反應導致在正極表面形成電子和離子絕緣的LiF,并導致過渡金屬離子溶解。此外,在高溫和高電池電壓下也可能產生此類酸性物質。為解決和消除過渡金屬離子溶解問題,廣泛使用了兩種策略。一是穩定正極材料的結構:(i)表面涂層可提供化學惰性層進行保護;(ii)陽離子和陰離子在體相層面的替代也已被廣泛應用,以調整晶體化學特性從而獲得更穩定的正極材料。另一種是添加電解質添加劑,以清除電解質中的水和/或HF雜質。
尖晶石結構正極材料
尖晶石結構的正極提供了一個消除鈷的機會。它們廉價且環保,由于存在三維(3D)鋰離子擴散通道,還具有優異的倍率性能。最廣泛研究的尖晶石正極是LiMn?O?。LiMn?O?的實際比容量約為120mAh/g,理論比容量為148mAh/g。LiMn?O?具有約4V(vs. Li/Li?)的電位,表現出優異的循環性能并在高倍率下保持容量。因此,LMO是高功率鋰離子電池的有前景的候選材料。這種尖晶石結構的主要問題來自于楊-特勒(John–Teller)效應和Mn3?在電解質中的溶解,導致不可逆的結構退化和容量衰減。
為改善電化學性能并彌補不足,其他元素被引入錳基尖晶石框架,形成LiM?.???Mn?.???O?(M = Al、Ti、Cr、Fe、Co、Ni、Cu和Zn)。例如,鎳替代的尖晶石材料,表示為LiNi?.?Mn?.?O?,被認為是一種有前景的高電壓尖晶石正極,因為它在~4.7V(vs. Li/Li?)通過Ni??/Ni2?雙重氧化還原工作,理論容量為147mAh/g,實際容量為125mAh/g。
多陰離子正極材料
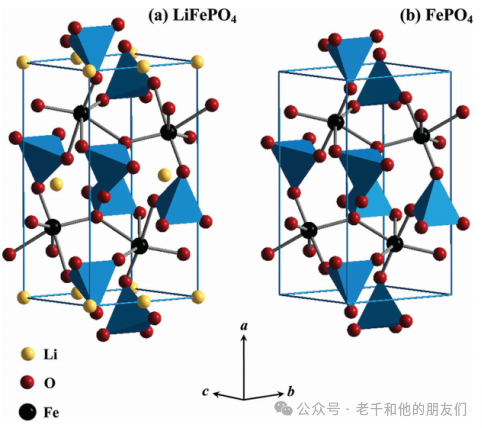
多陰離子正極由過渡金屬多面體和多陰離子基團組成的三維網絡構成。多陰離子正極由于共價鍵合的氧原子而具有高熱穩定性。最廣泛研究的多陰離子正極是具有有序橄欖石結構的磷酸鹽(LiMPO?,M = Fe、Mn、Co或Ni)。LiFePO?(LFP)是橄欖石磷酸鹽中研究最廣泛和發展最完善的材料。
如圖6所示,在充電過程中,由于脫鋰,LFP變成FePO?,而在放電過程中,由于鋰化,FePO?到LFP的可逆轉化發生。LFP表現出120-160mAh/g的實際比容量(理論容量:170mAh/g),3.4V(vs. Li/Li?)的平穩工作電壓和長循環壽命。然而,低內在電子導電性和慢的一維鋰擴散率限制了倍率性能。因此,LFP通常被碳涂覆或與其他導電劑混合以增強倍率能力。
圖6 (a) LiFePO4和(b) FePO4的晶體結構(磷的PO4環境已表示為四面體)
除了磷酸鹽外,硅酸鹽(Li?MSiO?)、氟磷酸鹽(LiVPO?F)和氟硫酸鹽(LiMSO?F)也被探索作為鋰離子電池的多陰離子正極。
無序巖鹽正極
無序巖鹽結構的正極,特別是陽離子無序巖鹽(DRX),是一種新型高能量密度正極材料。DRX中的三維主體結構相當穩定,因為隨機的陽離子分布消除了層間距的變化。基于DRX的氧化物和氧氟化物通常由地球上豐富的元素組成,同時具有無鈷化學性質、廣泛的組成空間和大的電荷存儲容量。一些DRX能夠提供超過300mAh/g和1,000Wh/kg的可逆容量和能量密度。金屬氧化物的電子導電性限制了正極材料的倍率性能,這可以通過各種策略來改善。還應注意的是,碳涂層可以改善基于V、基于Mn和基于Co的DRS氧化物的鋰擴散系數。
轉化型正極材料
與嵌入型正極不同,轉化型正極中的化學鍵在電化學反應過程中重復斷裂和重組。此外,由于多電子轉化反應,轉化型正極可以在每個金屬中心存儲多個鋰離子,從而導致比傳統正極高三到五倍的容量。在幾種類型的轉化材料中,金屬氟化物由于其高理論電位(3.55V [vs. Li/Li?],CuF?)和質量比容量(713mAh/g,FeF?)而最具前景。然而,過渡金屬氟化物由于過渡金屬-氟鍵的離子特性引起的大帶隙而具有較差的離子和電子導電性。通過利用各種碳復合材料、陽離子和陰離子替代以及納米結構化,已經取得了相當大的性能改進。
硫和氧氣
硫(S)提供1,675mAh/g的理論比容量和2.15V的平均電壓(相對于Li/Li+)。此外,低成本和豐富的儲量也賦予了硫顯著的競爭優勢。應當注意的是,硫是一種無鋰正極材料。Ji及其合作者通過在硫正極中引入高度有序的納米結構介孔碳主體,實現了高達1,320mAh/g的可逆容量,這引起了人們對鋰硫電池的熱情。
然而,硫正極存在一些固有缺點,如電絕緣的硫及其反應產物Li2S、液體電解質中可溶性鋰多硫化物的嚴重穿梭效應,以及鋰化/脫鋰過程中由體積變化引起的電極開裂和粉化。為解決這些問題,已經進行了多項努力,包括:(i)具有化學/物理限制功能的硫載體、中間層和改性隔膜;(ii)分子設計的活性材料;(iii)降低多硫化物溶解度的先進電解質;(iv)具有動力學加速功能的催化劑/氧化還原媒介;以及(v)負極保護。
氧氣是一種可以從空氣中獲取的氣態活性材料,能夠顯著降低工作電池的總重量。此外,O2和Li之間的轉化反應可以提供高儲電容量。典型的鋰空氣電池可以實現最高的質量能量密度,理論能量密度為3,500 Wh/kg,實際能量密度為950 Wh/kg。然而,電解質不穩定性、空氣電極的降解和鋰負極的不穩定性導致系統的循環性能差和能量效率低。升級電解質、保護金屬鋰負極和設計催化劑可以用來解決這些問題。
鋰離子電池的負極材料
提高鋰離子電池能量密度的另一個有效方法是尋求高容量負極材料。基于電化學鋰化/脫鋰機制,鋰離子電池中使用的負極大致分為三類:嵌入型負極、合金型負極和轉化型負極。
嵌入型負極——碳基材料
碳基材料,包括天然/合成石墨和軟/硬碳,由于其成本低、儲量豐富、脫鋰電位低(相對于Li/Li+)、鋰擴散性高、電導率高以及鋰化/脫鋰過程中體積變化小,成為鋰離子電池最可行的候選材料。值得強調的是,碳基負極使鋰離子電池在約40年前成為商業可行的產品,而且碳仍然是作為負極材料的最理想選擇。例如,傳統的石墨負極具有三維結構穩定、能量密度適中、理論質量容量為372mAh/g、理論體積容量為735 mAh/cm3以及低成本的優點。
迄今為止,石墨是商業鋰離子電池的主要負極材料。在充電過程中,來自電解質的Li+離子滲透到碳中并形成鋰/碳嵌入化合物,即LixC,這是一個可逆反應,每6個碳原子儲存1個鋰原子。需要注意的是,碳的質量容量比大多數正極材料高,但商業石墨的體積容量仍然較低,范圍在330到430 mAh/cm3之間。此外,還開發了納米結構碳基負極材料,如一維納米管、納米線、納米纖維、二維石墨烯和多孔碳基負極,以提高鋰離子電池的能量和功率密度。
插入型過渡金屬氧化物負極
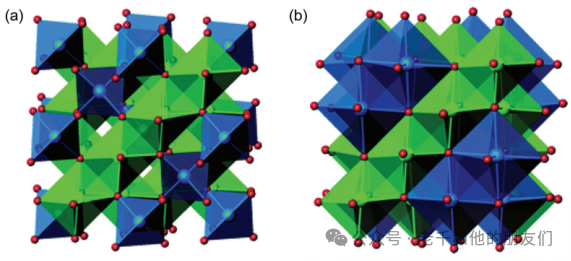
最經典的插入型過渡金屬氧化物負極是基于鈦的,例如Li4Ti5O12(LTO)和TiO2,其中氧化還原中心在鋰化時被還原為Ti3+,隨后在堿金屬陽離子脫出時重新氧化為Ti4+。尖晶石結構的LTO每單元可存儲多達三個Li+離子,經歷從尖晶石到巖鹽結構的可逆兩相反應,產生175mAh/g的理論容量,對應于化學式Li7Ti5O12。Li4Ti5O12和Li7Ti5O12的結構如圖7所示。
圖7 (a)尖晶石結構的Li4Ti5O12,其中藍色四面體代表Li,綠色八面體代表無序分布的Li和Ti;以及(b)巖鹽結構的Li7Ti5O12,其中藍色八面體代表Li,綠色八面體代表無序分布的Li和Ti。
LTO表現出較小的體積變化(~0.2%),被認為是零應變材料,這導致優異的循環性能。此外,高脫鋰電位(1.55V vs. Li/Li+)可以防止鋰枝晶的生長并保證基于LTO的鋰離子電池的安全性。此外,鋰離子擴散系數比石墨高一個數量級,確保了優異的倍率性能。
然而,LTO具有固有的較差電子導電性,這阻礙了倍率性能。通常采用兩種不同的方法來提高倍率性能:通過表面改性或離子摻雜來增強離子擴散和電子導電性,從而加速電荷轉移反應,以及通過設計納米結構LTO負極來減少鋰離子在體相中的擴散距離。
此外,各種結構的TiO2也被探索作為鋰離子電池的負極材料。由于沿[010]方向的特性平行通道,TiO2-B納米管或納米線被認為是用于鋰離子嵌入和擴散的最具吸引力的結構之一。
合金型負極
自從A. Dey證明鋰金屬可以在有機電解質中與其他金屬在室溫下進行電化學反應以來,鋰與不同金屬或半金屬元素和化合物之間的合金化反應被廣泛研究。第IV族元素,特別是Si和Sn,由于形成高容量富鋰二元合金和低工作電位(石墨除外),成為合金化反應的主要研究對象。Si和Sn的理論容量分別為3,579和994mAh/g,顯示出對下一代鋰離子電池的潛力。
硅和硅基化合物
純硅負極與鋰的反應機理可解釋如下:
放電過程:Si(結晶態) + xLi+ + xe- → LixSi(非晶態)
(3.75-x)Li+ + (3.75-x)e- → Li15Si4(結晶態)
?
充電過程:Li15Si4(結晶態) + y → Si(非晶態) + yLi+ + ye- + Li15-ySi4(殘留)
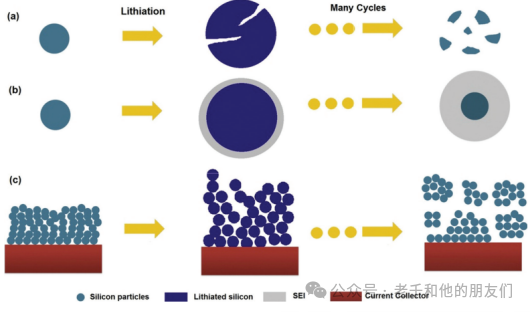
在第一次放電的所有階段都觀察到上述兩相反應,并在完全形成二元合金(Li15Si4)后完全消失。因此,在隨后的循環中只觀察到單相反應。然而,硅在鋰化/脫鋰過程中伴隨著劇烈的體積膨脹(300%-400%)和巨大的應力產生,導致一系列嚴重的破壞性后果:(i)由于重復的充放電過程中逐漸增強的粉化,電極的結構完整性受到破壞;(ii)由于界面應力的存在,電極與集流體之間的連接斷開;以及(iii)在固體電解質界面(SEI)層持續形成-破裂-重新形成過程中,鋰離子持續消耗(圖8)。
圖8 硅電極的三種不同失效機制:(a)電極粉化,(b)SEI層的持續破裂和重新生長,以及(c)整個電極的崩塌。
已經開發了幾種設計策略來開發納米結構硅負極并克服這些問題,因為(i)納米結構硅負極可以在電池操作中避免機械斷裂,(ii)納米結構硅與納米結構碳的結合,如典型的蛋黃殼或管中線結構,可以確保電池操作過程中高效的電子和離子傳輸,以及(iii)納米結構硅與碳的共價鍵合可以減少由活性材料與集流體分離引起的意外斷連。
此外,一氧化硅(SiO)也被視為鋰離子電池的負極候選材料。SiO在充放電過程中的絕對體積變化比硅小。此外,鋰插入形成的幾種含氧化合物有望在緩解體積變化方面發揮基質作用。
錫(Sn)和錫基化合物
1997年,日本富士膠片Celltec有限公司宣布其Stalion電池使用錫基非晶氧化物作為負極。該材料由Sn-O作為鋰插入的活性中心和其他形成玻璃的元素組成,可提供>600mAh/g的質量容量,鼓勵更多研究人員從事錫基材料研究。
錫基氧化物材料的基本反應機理可表述如下:
SnO + 4Li+ + 4e- → Sn + 2Li2O
Sn + 4.4Li+ + 4.4e- Li4.4Sn + 2Li2O
SnO2 + 4Li+ + 4e- → Sn + 2Li2O
Sn + 4.4Li+ + 4.4e- Li4.4Sn + 2Li2O
從Sn到完全鋰化的Sn(Li4.4Sn)的轉變過程中,體積變化約為260%,這是錫負極容量急劇衰減的主要原因。在充放電過程中,一種鋰錫相以另一種為代價而生長,不同鋰錫合金之間的晶格參數和結構差異導致應力/應變的積累和釋放,引起顆粒開裂、SEI重新形成、與集流體連接喪失和電極失效,導致循環性能的快速惡化。已經做了大量努力來克服這些問題,包括設計和探索具有不同尺寸、形狀和孔隙率的錫基化合物。
多年來,研究人員報道了各種基于錫的氧化物或氧化物玻璃,它們具有高容量、優異的循環穩定性和卓越的倍率性能。然而,其初始庫侖效率仍需提高。例如,Dahn等人研究了基于錫的化合物的電化學性能以及鋰與不同錫氧化物玻璃反應過程中的結構變化,證明了在首次循環中存在顯著的不可逆容量損失(200-700mAh/g)。
除了使用錫氧化物作為非活性分散劑外,另一種方法是從基于錫的合金開始。2005年,索尼發布了采用納米結構Sn-Co-C基負極的Nexelion 14430型電池。索尼聲稱,與傳統鋰離子電池相比,其體積容量可以提高30%以上。
2011年,索尼宣布推出另一代Nexelion電池(18650型電池),在2.0V-4.3V電壓范圍內,容量為3.5 Ah,體積能量密度為723 Wh/L。此外,受索尼Sn-Co-C負極成功的鼓舞,研究重點轉向錫-過渡金屬合金,如Sn-Cu、Sn-Ni、Sn-Fe和Sn-Co合金。
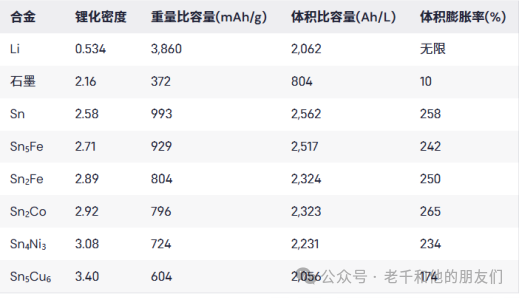
表1總結了Sn、Sn5Fe、Sn2Fe、Sn2Co、Ni3Sn4和Cu6Sn5形成Li4.4Sn+M時的鋰化密度、質量容量和體積容量,并與鋰金屬和石墨進行了比較。
表1錫、Sn?Fe、Sn?Fe、Sn?Co、Ni?Sn?和Cu?Sn?合金的鋰化密度、重量比容量和體積比容量
轉化型負極
轉化是一種可逆的電化學反應(通常稱為置換反應),其中過渡金屬化合物(MXy,X = P、S、O、F或Cl)在電化學過程中被破壞并隨后還原為金屬(M0)。當用作鋰離子電池的負極時,過渡金屬化合物(如氧化物、磷化物、硫化物和氮化物(MxNy;M = Fe、Co、Cu、Mn、Ni和N = O、P、S和N))的電化學反應機理是過渡金屬的還原(氧化)以及鋰化合物(LixNy;N = O、P、S和N)的組成(分解)。
電化學轉化反應可描述如下:
MxNy + ze- + zLi+ M + zLiyNy,
其中M = Fe、Co、Cu、Mn、Ni,N = O、P、S和N。
基于轉化反應的過渡金屬氧化物(TMOs),包括Fe2O3、Co3O4、MnO、CuO和NiO,是鋰離子電池的典型負極材料。這些化合物的優點是在充放電過程中體積變化較小,且由于過渡金屬氧化物轉化為過渡金屬和鋰氧化物過程中每個過渡金屬的多電子轉移反應,它們具有高容量(Fe2O3約1,000mAh/g)。然而,基于轉化反應的TMO負極的工業化受到其較差的電導率和循環性能的阻礙。
與合金負極類似,轉化型負極也存在材料顆粒級別的粉化、不穩定的SEI層以及電極級別的形態和體積變化等問題。為了實現轉化氧化物的循環,需要采用納米設計策略實現多固相的相互轉化。
金屬鋰負極
除了上述金屬和化合物外,金屬鋰也可用作負極材料。事實上,金屬鋰在鋰電池研究初期就已被使用,包括20世紀70年代由埃克森美孚的Stanley Whittingham開創的第一個可行的鋰離子電池。金屬鋰是終極負極選擇,因為它具有最高的理論容量(3,860mAh/g或2,061 mAh/cm3),低密度(0.59 g/cm3)和最低的電化學電位(相對于標準氫電極為-3.04V)。
此外,鋰金屬負極對于鋰硫和鋰空電池系統不可或缺,這兩種系統都正在被廣泛研究用于下一代能量存儲應用。與石墨負極的嵌入/脫嵌機制不同,鋰金屬負極中發生的是金屬鋰與鋰離子之間的轉化反應。
一旦負極被鋰金屬取代,使用含鋰過渡金屬氧化物作為正極的鋰離子電池的比能量可從280提高到約440 Wh/kg,而鋰硫和鋰空系統可進一步將比能量提高到約650和約950 Wh/kg。鋰空電池的體積能量密度接近汽油。
然而,不可控的鋰枝晶生長導致短壽命和災難性的安全隱患,限制了鋰負極的實際應用。已采用一些有效策略來克服這些挑戰,包括電解質改性、引入保護層、納米結構負極和膜修飾。盡管實驗結果令人鼓舞,但由于鋰金屬的活潑性質,在實際電池應用中仍有很長的路要走。
電解質
電解質在所有電化學設備中都是無處不在且不可或缺的。電解質的作用是作為在正極和負極之間傳遞電荷的媒介。電解質與其他組件(包括正極、負極和隔膜)緊密接觸。界面,主要是電解質與電極之間的界面,通常決定了鋰離子電池的性能。因此,電解質必須對正極和負極表面都表現出穩定性。
鋰離子電池理想的電解質應滿足以下要求:高離子電導率、寬電位范圍內的電化學穩定性、化學穩定性、熱穩定性、成本效益、簡單的制備過程、低毒性和環保性。此外,電解質的電化學工作窗口應被修改以開發高電壓正極和低電壓負極材料。
鑒于電極-電解質界面對電池性能的重要性,電極/電解質界面,即固體電解質界面(SEI)和正極-電解質界面(CEI),它們分別通過電解質在負極/電解質和正極/電解質界面的電化學分解形成,將首先被簡要介紹。
電極/電解質界面
一般來說,電極-電解質界面可以被視為覆蓋在電極顆粒上的薄膜,由電解質的分解反應形成,保護活性材料免受后續降解機制的影響。
負極-電解質界面,稱為固體電解質界面(SEI),影響鋰離子從溶劑化相進入固相的過程,構成了大多數電極材料鋰化過程的速率限制步驟。
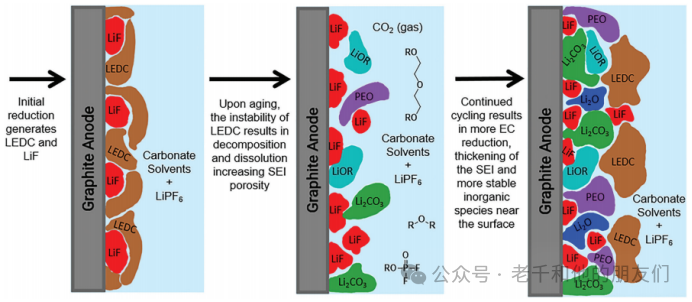
負極材料的電化學工作電位低于鋰基電池電解質中常用的有機碳酸鹽的還原電位(約為~1V vs. Li/Li+)。在電池充電過程中,電解質的電化學還原發生并在負極表面產生鈍化SEI層。初始SEI組分的分解反應以及老化過程中SEI組成的觀察變化,導致了以下SEI演變機制的提出(圖9)。
圖9 石墨負極上初始SEI形成的示意圖,酸介導的熱分解反應對SEI結構的影響,以及電解質的進一步還原導致SEI增厚的過程。
SEI是鋰離子導體但電子絕緣體,導致SEI在一定厚度下停止生長。穩定的SEI層由于表面鈍化效應,允許高庫侖效率和負極的長期穩定性。然而,由于鋰化和脫鋰過程中重復的大體積變化,電極/電解質界面顯著移動和變化,使得為高容量電極材料維持穩定的SEI變得極具挑戰性。
在其他電池化學體系中,如石墨和硅負極電極,SEI形成過程僅涉及電解質在電極上的電化學還原分解,因為石墨和硅具有化學穩定性。鋰金屬上SEI的形成包括化學和電化學反應。因此,盡管鋰金屬上形成的SEI與石墨和硅電極上形成的SEI具有相似功能,但兩類SEI之間的基本區別不應被忽視。
與SEI不同,CEI的研究較少,因為大多數正極的工作電位與商業碳酸鹽電解質的熱力學穩定窗口沒有太大偏差。隨著高電壓鋰離子電池的發展,當截止電壓超過電解質的氧化穩定性時,電解質將在正極表面氧化和分解。由于高電壓操作的要求,對CEI的理解變得越來越重要。然而,CEI的精確結構和組成仍存在爭議,其結構和化學性質如何影響電池安全性尚未被完全理解。對于不同正極材料的CEI形成,通常提出交換反應和親核反應機制。
有機電解質
有機電解質或非水系電解質,通常由鋰鹽溶解在有機溶劑或溶劑混排物中組成,是鋰離子電池中最常用的電解質,因為它們具有高離子電導率和對多孔隔膜及電極的優良潤濕性。
當第一個鋰嵌入式正極材料,即TiS?被發明時,由于TiS?的工作電位適中(<3.0V vs. Li/Li?),處于醚類穩定性范圍內,因此使用了醚類電解質(二氧戊環中的LiClO?)。當層狀過渡金屬氧化物被用作高壓正極材料時,電解質從醚類轉向酯類,因為酯類對正極具有良好的氧化穩定性,且能溶解多種鋰鹽以提供良好的離子電導率。
在所有酯類電解質中,碳酸丙烯酯(PC)和碳酸乙烯酯(EC)是兩種常用溶劑。1990年索尼商業化的第一代鋰離子電池使用無定形碳作為負極,采用了基于PC的電解質。另一方面,當石墨用作無定形碳的替代品時,開發了基于EC的電解質。此外,LiPF?因其平衡的性能而被廣泛用作有機電解質中的鋰鹽,適用于不同的電池環境。最終,大多數商業鋰離子電池使用LiPF?溶解在EC和線性碳酸酯(如碳酸二甲酯(DMC)、碳酸二乙酯(DEC)或碳酸甲乙酯(EMC))的混排物中。
此外,電解質添加劑通過引導SEI形成、提高電解質離子電導率、增加LiPF?熱穩定性以及保護電極免受溶解和過充等方式,能有效提高鋰離子電池的性能和循環壽命,已被廣泛應用于電解質配方中。
電解質添加劑通常是無機化合物和具有各種官能團的有機化合物,如不飽和碳鍵、含硫組分、含鹵素組分和其他組分。一般來說,對于負極,電解質添加劑在高于電解質溶劑的電位下還原并鈍化電極表面,防止電解質溶劑進一步還原;對于正極,電解質添加劑預期在溶劑之前氧化并覆蓋電極表面,防止電解質的氧化分解。
考慮到不同類型的電極材料,電解質添加劑的選擇可能會改變。例如,含氟和含磷電解質添加劑是能夠改善高壓正極材料性能的有前途的物質,而丁基砜可以改善LiFePO?的低電子電導率及其在低溫下的倍率性能。
水系電解質
相比易燃的有機電解質,使用具有高離子電導率的水系電解質在成本、環境和安全性方面具有優勢。因此,Dahn小組首先使用硝酸鋰水系電解質,在5M LiNO?電解質中采用VO?負極和LiMn?O?正極構建了水系鋰離子電池,平均工作電壓為1.5V,能量密度約為55Wh/kg。然而,可能發生的H?或O?析出的副反應以及電極材料與水或溶解的O?的溶解/副反應常導致低庫侖效率和差的循環性能。此外,各種金屬氧化物正極,如LiCoO?、LiMn?O?或NMC,在水系電解質中表現出pH敏感性。
最近,通過在水中添加高濃度的不同鹽類開發了"水中鹽"電解質。Wang小組首先證明,通過使用合適的陰離子(N(SO?CF?)??或TFSI?)操控Li?離子溶劑化結構,降低水的電化學活性,可以提高水分解的陽極和陰極極限。首次充電過程中在電極和電解質之間形成的界面將電化學窗口擴展到約3.0V。應該注意的是,水中鹽電解質使用高濃度鹽阻礙了實際應用。因此,研究工作應致力于降低電解質成本,同時不犧牲SEI形成能力和增加水活性的獨特特性。
離子液體
離子液體(ILs)由陽離子和陰離子組成,不含任何溶劑,具有低熔點和良好的離子電導率。不同的陽離子,如咪唑鎓、季銨、吡咯烷鎓和哌啶鎓,與陰離子如PF??、BF??和雙(三氟甲磺酰)亞胺(TFSI?)結合,為電解質應用提供不同的性能。近來,室溫離子液體因其寬廣的電化學操作范圍、無揮發性和改善的熱穩定性而受到更多關注。然而,與商業有機電解質相比,它們也存在較差的功率性能。因此,在商業化之前還有很多工作要完成。
固態電解質
傳統鋰離子電池因使用高度易燃、熱穩定性低和閃點低的有機液體電解質而存在嚴重安全問題,容易導致火災事故和爆炸。因此,正在開發固態電解質(SSEs)以追求具有高能量密度和改善安全性的下一代能源存儲設備。
與液體電解質相比,固態電解質不揮發、不易燃且具有高熱穩定性,使其適用于廣泛的操作溫度范圍。固態電解質提供了一個物理屏障層來分離正極和負極,并防止在高溫或沖擊下的熱失控。此外,固態電解質可以通過有效抑制鋰枝晶的形成,實現鋰金屬負極的成功利用。
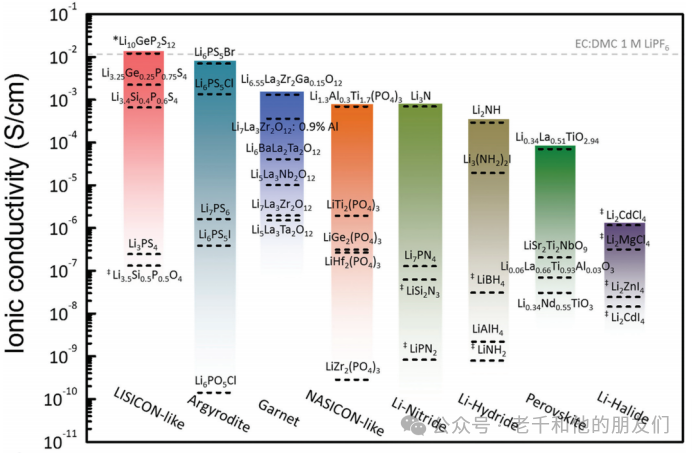
為滿足商業要求,高離子電導率、良好的機械性能和與電極的出色界面穩定性是固態電解質最基本的要求。因此,已開發了幾種類型的固態電解質,包括無機固態電解質(鈉超離子導體型、鈣鈦礦型、石榴石型和硫化物型)、聚合物和復合固態電解質以及薄膜固態電解質。圖10比較了選定無機固態電解質與典型有機液體電解質的鋰離子電導率。一些結構家族,如LISICON類、銀黃晶石和石榴石,在室溫下可實現10?2至10?3S/cm范圍內的高離子電導率,而每個結構家族內的Li?電導率可能存在高達5-6個數量級的巨大差異。
圖10無機固態電解質和典型有機液態電解質的鋰離子電導率。
總結與展望??
本文簡要介紹了鋰離子電池以及超越鋰電池的發展,并總結了鋰離子電池正極、負極和電解質材料的發展。
未來鋰離子電池和其他可充電電池的發展應考慮以下幾個方面:
i. 應通過探索新型材料并擴展我們對結構-成分-性能-表現關系的基本理解,進一步開發高能量密度、高容量和高電壓的正極材料。值得強調的是,追求高容量正極材料是提高電池能量密度最有效的方法,而高電壓正極材料可以降低鋰離子電池模塊的復雜性。對于負極材料,混排多種材料以追求互補特性應該是一個發展方向。
ii. 快速充電或超快速充電(XFC)是提升用戶充電體驗的關鍵驅動力和長期戰略目標。根據美國先進電池聯盟(USABC)的標準,快速充電是指在15分鐘內使電池達到80%的充電狀態,這意味著電池組可以以4C或更高的速率充電至80%的SOC。雖然大多數商業鋰離子電池的最大充電速率僅限于3C,但應探索快速充電的正極和負極材料。
iii. 對成分和/或結構演變與電池性能之間關系的全面理解仍然有限。過去十年里,創新的實驗和建模方法在多尺度上研究復雜材料的應用呈爆炸性增長。然而,先進的表征技術(即原位和同步操作工具、低溫電子顯微鏡)和理論/計算分析對于電池材料的進一步發展仍然是必需的。
iv. 高溫和低溫環境都會嚴重影響電池容量和使用壽命。在高溫環境中,鋰離子電池可能產生熱失控,導致短路、燃燒、爆炸和其他安全問題。另一方面,在低溫下,鋰離子電池中可能出現鋰枝晶,導致短路、啟動失敗和其他操作故障。因此,高效的電池熱管理系統(BTMS)對于最大化電池模塊/組的壽命具有至關重要的意義。
v. 鋰離子電池的可持續回收技術已部分建立;然而,回收技術遠未成熟,改進鋰離子電池的回收技術是一項持續的努力。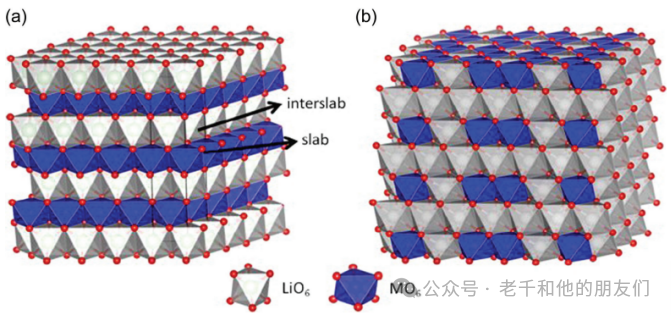
-
鋰離子電池
+關注
關注
85文章
3373瀏覽量
78998 -
電極
+關注
關注
5文章
842瀏覽量
27840 -
電池
+關注
關注
84文章
11070瀏覽量
134893
原文標題:鋰離子電池技術解讀 | 入門指南
文章出處:【微信號:bdtdsj,微信公眾號:中科院半導體所】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄





 工商網監
工商網監
評論