 濃度極化誘導相變穩定聚合物電解質中的鋰鍍
濃度極化誘導相變穩定聚合物電解質中的鋰鍍
背景介紹
鋰金屬負極具有3860 mAh g-1的理論容量,以及超低的電極電位(-3.04 V),因此有望極大提高電池能量密度。然而,液體電解質中的濃差極化促進了金屬沉積過程中晶須的生長,導致鋰沉積不均勻,形成粗糙的形貌,如苔蘚狀和樹枝狀的鋰。這種不均勻沉積不僅導致電極表面積大,促進與電解質發生副作用,降低庫侖效率(CE)和循環壽命,而且還存在內短路、熱失控等安全隱患,尤其是與傳統的易燃液體電解質(如醚類、碳酸鹽)匹配時。聚合物電解質雖然不能完全解決鋰金屬電池(LMBs)中的安全問題,但由于其比液體電解質的熱穩定性更高,因此有助于提高LMBs安全性。遺憾的是,聚乙烯氧化物(PEO)電解質的楊氏模量,通常在20-70 MPa范圍內,遠低于有效抑制Li晶須所需的1 GPa閾值。因此,Li晶須在聚合物電解質中快速生長。當引入增塑劑來增強離子電導率時,會進一步軟化聚合物電解質,晶須生長變得更加嚴重。解決這一挑戰需要對動態的Li金屬/聚合物電解質界面有基本的理解,例如Li+濃度如何在Li負極表面演變,以及Li負極如何與固體電解質相互作用。
二、正文部分
1、成果簡介
近日,哥倫比亞大學楊遠教授,閔瑋教授和Qian Cheng,通過受激拉曼散射顯微鏡證明,固體聚合物電解質中的濃差極化可以誘導聚乙烯氧化物(PEO)電解質發生相變,在鋰/電解質界面形成一個富PEO但貧鹽/增塑劑的新相。與本體聚合物電解質(《1 MPa)相比,新相具有高得多的楊氏模量(~1-3 GPa)。因此,PEO電解質的組成應在相圖中單相區和兩相區的邊界附近,這樣施加的電流可以誘導形成機械剛性的富PEO相來抑制鋰晶須。具有濃差極化誘導相變的LiFePO4/PEO/Li電池可以可逆循環100次,而沒有這種相變的電池在10次循環內失效,證明了該策略的有效性。該研究以題目為“Stabilizing lithium plating in polymer electrolytes by concentration-polarization-induced phase transformation”的論文發表在國際頂級期刊《Joule》上。

2、研究亮點
本工作利用具有高時間分辨率、成像速度和靈敏度的受激拉曼散射(SRS)顯微鏡研究了固體聚合物電解質(SPE)與電極的相互作用。結果表明,濃差極化并沒有促進晶須的生成,而是降低了鋰/電解質界面的鹽濃度,使單相PEO電解質轉變為兩相PEO電解質。這導致在鋰/電解質界面形成了模量為~1-3 GPa的機械剛性富PEO相,對應的剪切模量為0.36-1.06 GPa(圖1B)。這么的高模量抑制了晶須的生長,使鋰沉積均勻。因此,電解質成分應位于PEO-鹽-增塑劑相圖中單相區和兩相區的邊界處,這樣通過小電流誘導形成輕微的濃差極化,就可以在金屬鋰表面獲得機械剛性的富PEO相,使金屬鋰鈍化。具有濃差極化誘導機制的LiFePO4(LFP)/PEO/Li電池能夠穩定循環超過100次。
3、圖文導讀
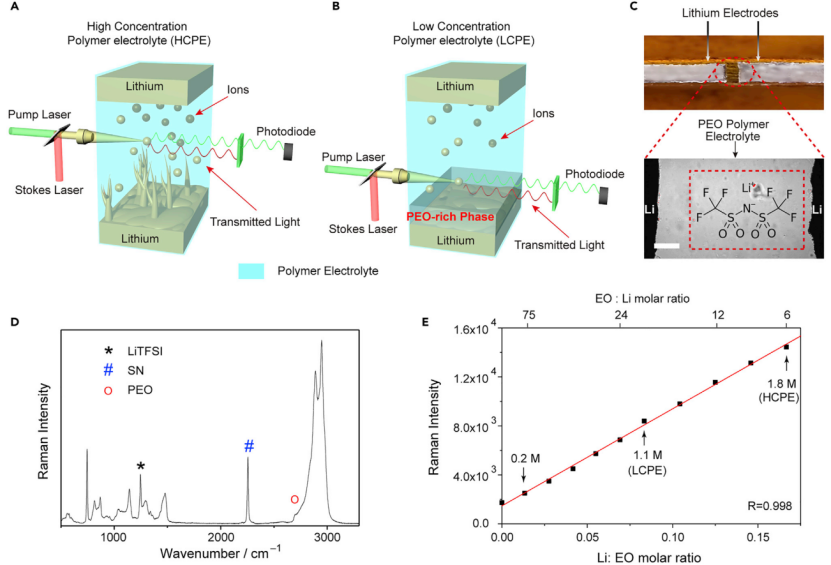
【圖1】(A和B)Li/Li電池中高濃度聚合物電解質(HCPE)和低濃度聚合物電解質(LCPE)的SRS成像示意圖。(C)Li/PEO/Li電池的亮場圖像。上圖為電池結構,下圖為光學顯微鏡放大后的圖像,比例尺為100 mm。(D)LCPE的拉曼光譜。LCPE對應的組成為EOSN=122.64(摩爾濃度)。(E)1245 cm-1處LiTFSI峰的拉曼強度圖隨PEO電解質中Li:EO比的演變,顯示出良好的線性關系。1.1 M和1.8 M LiTFSI的點分別對應LCPE和HCPE。
自制了鋰對稱電池,并通過SRS顯微鏡研究Li/PEO電解質相互作用(圖1C)。在該電池中,PEO電解液填充在兩塊鋰之間的空隙中,所有組件被夾在兩塊玻片之間,用環氧樹脂密封。PEO電解質以雙(三氟甲磺酰)亞胺鋰(LiTFSI)為鹽,琥珀腈(SN)為增塑劑,以增強離子電導率,使其可在室溫(RT)下運行。圖1D的LCPE拉曼光譜顯示,在1245 cm-1(C-F拉伸)、2250 cm-1(C≡N拉伸)和2800 cm-1(C-H的拉伸振動)附近的峰分別為LiTFSI、SN和PEO的拉曼特征峰(圖1D)。由于電解質滿足電中性要求,因此[Li+]可以被認為等于[TFSI-]。測量[TFSI-]可以用來表示局部的[Li+]。而TFSI-的拉曼強度與其濃度成正比,因此可以將拉曼信號轉換為化學濃度(圖1E)。
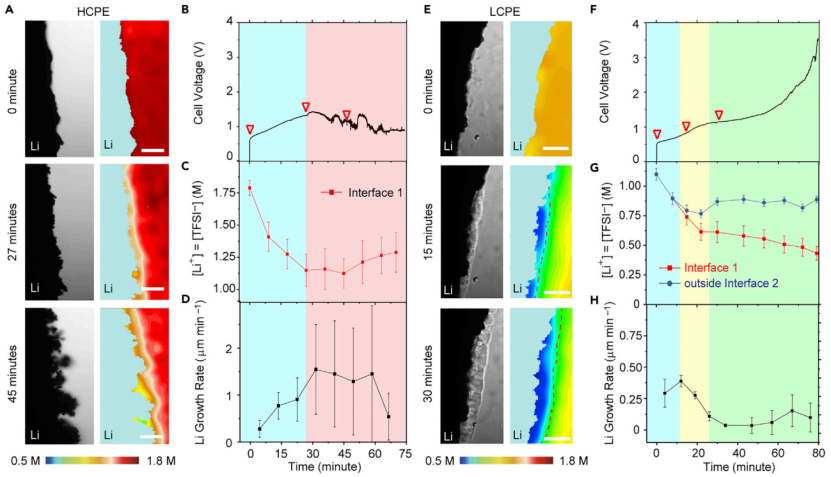
【圖2】(A-D)HCPE中鋰晶須的生長。(A)[Li+]=[TFSI-]三個代表性階段的亮場及對應的SRS圖像。(B)(A)中Li/Li電池的電壓曲線。(C)[Li+]=[TFSI-]隨時間的變化,(D)鋰生長速率(v)隨時間的變化。界面1為鋰電極與PEO電解質的邊界。(E-H)鋰在LCPE中的生長。(E)[Li+]三個代表性階段的亮場圖像和SRS圖像。(F)(E)中Li/Li電池的電壓曲線。黃色的陰影對應富PEO相的出現,綠色的陰影表示富PEO相已經覆蓋整個鋰表面。(G)[Li+]=[TFSI-]隨時間的變化,(H)鋰生長速率(v)隨時間的變化。界面2為富PEO相與各向同性體相聚合物電解質之間的邊界,如圖(E)中SRS圖像中的虛線所示。比例尺為50 mm。兩個電池的電極距離均為500mm。在室溫下,對兩個電池施加0.5 mA cm-2的電流密度。
以EOSN=122.64和122.64,分別制備了高濃度聚合物電解質(HCPE)和低濃度聚合物電解質(LCPE),并進行了研究,HCPE對應的LiTFSI和SN濃度分別為1.8 M和2.4 M, LCPE對應的LiTFSI和SN濃度分別為1.1 M和2.9 M。兩種情況下SN與PEO的重量比均固定在40%。
對于HCPE(圖2A),施加0.5 mA cm-2的電流時,鋰表面的[Li+]([Li+]0 μm)從t=0時的1.8 M逐漸消耗到t=27 min時的1.2 M,之后[Li+]0 μm在穩定在~1.2 M(圖2B和2C)。同時,在t=0的鋰生長速率(v)從0.27±0.18 μm min-1迅速增加到t=27 min的0.9±0.46 μm min-1(圖2D)。之后,v在剩余的時間里急劇增加到~1.5 μm min-1,導致了97%的超高孔隙率,表明HCPE不能抑制晶須的生長。這些結果表明,如果不發生相變,Li/聚合物電解質界面的離子濃差極化促進了晶須的生長。
圖2E和2F的SRS圖像和明場(BF)圖像顯示,與HCPE中觀察到的行為相反,LCPE中的濃差極化導致鋰/電解質界面發生相變過程,這反而能抑制鋰晶須的生長。首先,在SRS中,新相出現在藍色區域,在BF中表現為粒狀區域。從Li/電解質界面(界面1)上的[LiTFSI]和新相與各向同性體相電解質界面(界面2)外的[LiTFSI]對比可以看出,富PEO相的[LiTFSI]比相鄰各向同性體相電解質中的[LiTFSI]低得多(圖2G)。這一差異在t=15 min時為0.79/0.74 M,在t=30 min時為0.87/0.61 M,在t=79 min時為0.89/0.43 M。
這一新相的出現能夠有效地抑制鋰晶須的生長。雖然在t=0時觀察到鋰晶須生長,v為~0.3 μm min-1(87%孔隙率),但在t=30 min時,富PEO相初步形成后,v迅速下降到0.048 μm min-1(圖2H),相當于孔隙率為16%。在這一階段,富PEO相逐漸在金屬鋰表面形成。新相完全覆蓋Li金屬表面后,30 ~ 63 min的平均v僅為0.044 μm min-1,孔隙率為9.2%,約為HCPE的三十分之一,表明鋰沉積致密均勻。這種行為在濃差極化過程中不僅是自發的,而且是自增強的。例如,如果鋰晶須在某一位置生長迅速,局域電流密度會增加,導致濃差極化更大,從而形成更厚的富PEO相,從而抑制晶須的生長。
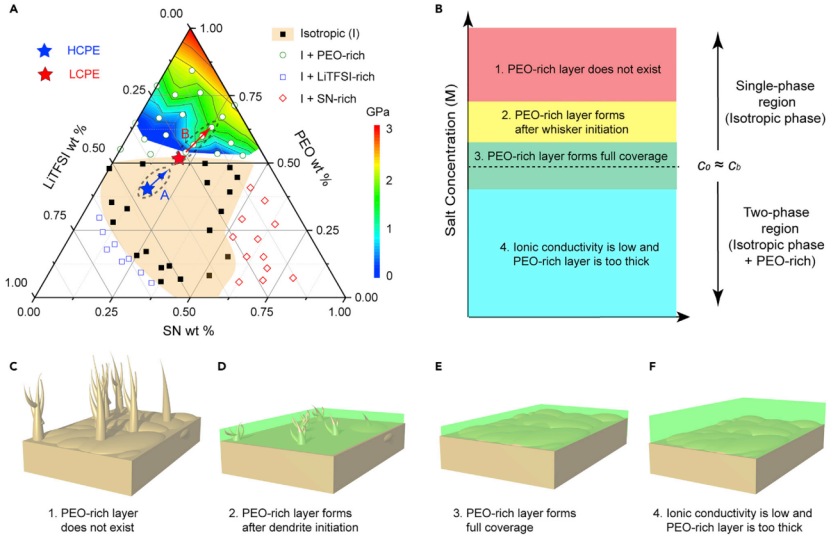
【圖3】(A)PEO-LiTFSI-SN三元相圖。橙色代表各向同性單相區域,其他區域代表兩相區域。三角形頂部的彩虹色區域代表相圖中相應組分形成的富PEO相楊氏模量分布。(B)當施加電流時,聚合物電解質中初始鹽濃度影響富PEO層厚度和覆蓋面積的示意圖。(C)當c0較高時,沒有形成富PEO層,容易形成鋰晶須(c0cb)。(D)c0略高于cb,因此晶須通常比富PEO層更早形成。這導致了鋰晶須生長的部分鈍化。(E)c0cb時,富PEO相對金屬鋰的鈍化效果最好。(F)c0《cb時,它具有較低的離子電導率,可能導致厚的富PEO層。
在排除電解質分解的可能性后,在高通量SRS的幫助下構建了PEO-LiTFSI-SN三元相圖(圖3A),以研究相隨成分的演化。相圖由中間(橙色)的單相各向同性區(I區)和角落處的三個兩相區(白色區)組成。圖3A顯示,HCPE的組成位于I區中心。當施加電流時,鋰金屬表面的[LiTFSI]逐漸降低(圖3A中的路徑A)。由于HCPE中鹽濃度較高,濃極化過程中Li/電解質界面處的電解質成分保持在I區,沒有發生相變。
相比之下,LCPE的組成非常接近單相I區和兩相區的邊界(cb),容易發生濃差極化(圖3A中的路徑B和3E),因為形成兩相比保持單相在熱力學上更穩定。由于較大的離子濃度梯度,較低的t+可以在較早的時間觸發相變。這種相變不僅使新相的[LiTFSI]降低,而且使[SN]降低,將SN排斥到各向同性電解質中。
在EOSN=82.64的Li/Li電池中,鹽濃度略高于cb。鋰沉積開始后,鋰晶須先生長,未觀察到富PEO相。而當鋰表面鹽濃度逐漸降低至低于cb時,富PEO相出現并增厚,抑制了鋰晶須的生長。但由于鋰晶須最初呈針狀,導致富PEO相不能完全覆蓋在鋰表面,破壞了保護的有效性。最后,測試了濃度為0.52 M LiTFSI和2.6 M SN的聚合物電解質,該聚合物電解質位于兩相區深處。這種固體電解質具有較低的離子電導率,無法正常運行。
從以上四種情況可以得出結論,富PEO層來源于RT時或近RT時的固相轉變,其形成高度依賴于PEO電解質中的鹽濃度(圖3B)。如果初始鹽濃度(c0)在單相區較深處,則HCPE(c0cb)時,不太可能形成富PEO相(圖3C)。當c0略高于cb時,晶須可能在富PEO相形成之前就開始形成,因此不能完全抑制晶須生長(圖3D)。如果c0接近cb(LCPE),即使很小的電流也能形成富PEO相,在短時間內完全覆蓋鋰表面,有效抑制晶須(圖3E)。如果c0《cb,聚合物電解質的離子電導率通常較低。此外,富PEO的相很容易在金屬鋰表面形成,但它可能太厚而不能導電(圖3F)。
基于以上分析,提出了LMBs聚合物電解質的設計原則:電解質成分應位于PEO-鹽-增塑劑相圖中單相區域和兩相區域的邊界處,這樣只需要很小的電流就會導致鹽濃度降低,并在金屬鋰表面形成機械強度高的富PEO相。
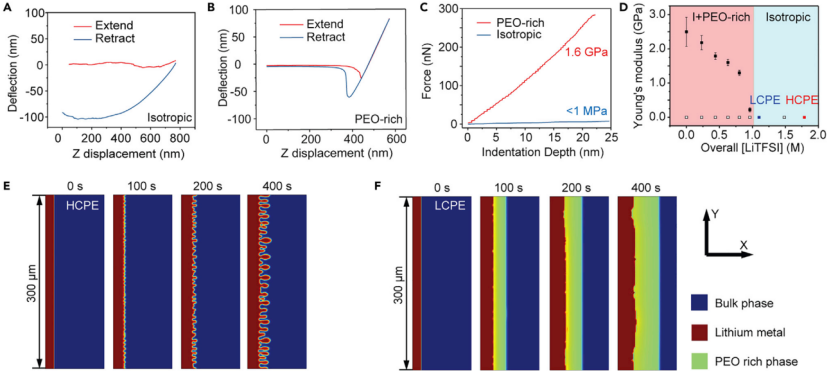
【圖4】(A)各向同性體相電解質(I區,HCPE)和(B)富PEO相的AFM拉伸和收縮力曲線。(C)(A)和(B)樣品對應的力-壓痕曲線。(D)不同鹽濃度下富PEO相的楊氏模量。(E)高濃度(2 M鹽)和(F)低濃度(1 M鹽)固體聚合物電解質在 0.5 mA cm-2下鋰電沉積的相場模擬。
富PEO相對鋰晶須的有效抑制是由鋰/電解質界面相變過程中的機械力-化學耦合引起的。為了驗證這一點,使用AFM測量了不同組成的SPE楊氏模量,包括各向同性相(I區)和富PEO相,并將結果疊加在圖3A的相圖上。對于HCPE(1.8 M LiTFSI, 2.4 M SN),接近曲線顯示,當尖端壓入電解質時,懸臂梁沒有變形,說明電解質非常柔軟(圖4A)。針尖縮回時檢測到大的懸臂撓度,證實針尖被壓在HCPE內部,電解質有粘性。圖3A中I區內的所有SPE和兩相區域中SPE的各向同性相顯示出相似的結果(楊氏模量《1 MPa),表明它們不能抑制鋰晶須的生長。
相反,SPE中形成的富PEO相表現出不同的力曲線。樣品整體組成為0.6 M LiTFSI, 3.3 M SN,其中富PEO相包含0.52 M LiTFSI和2.6 M SN(圖4B)。采用Sneddon模型,對應的模量為1.6 GPa(圖4C),壓痕硬度為~410-780 MPa。材料的壓痕硬度通常是其抗拉強度的3倍,這意味著富PEO相的抗拉強度為~137 MPa。這遠遠超過了金屬鋰的硬度(~7-43 MPa)和屈服強度(0.6-1.3 MPa),所以在鋰沉積過程中金屬鋰的蠕變是不可避免的,導致鋰晶須的生長受到抑制。而體相PEO電解質的模量《1 MPa,與金屬鋰的硬度和屈服強度相近或較小,因此不能抑制鋰晶須的生長。
為了進一步了解Li/電解質界面上的機械力-化學耦合作用,本研究利用原子力顯微鏡(AFM)系統地測定了PEO電解質中富PEO相的機械性質。圖4D顯示,當富PEO相的[LiTFSI]分別降至0.80和0.44 M時,富PEO相的模量迅速上升至1.2 ~ 1.8 GPa。進一步研究I+富PEO兩相區的富PEO相楊氏模量(圖3A)可以看出,當[LiTFSI]小于0.8 M時,模量通常在1 GPa以上。由于純PEO的楊氏模量為5-7 GPa,因此,在有限鹽/增塑劑的半晶化富PEO層中,其楊氏模量可以達到1-2 GPa。上述結果表明,濃差極化誘導的鋰沉積穩定機制是有效的。
相場模擬結果也證實了上述抑制機制的合理性,該模擬考慮了機械力-化學耦合。基于上述結果,模擬假設在[LiTFSI]《0.85 M時形成富PEO相,富PEO相的楊氏模量為1.6 GPa,而[LiTFSI]0.85 M區域的模量為1 MPa。由于在HCPE中沒有形成新相,鋰晶須在軟的各向同性體聚合物電解質中快速生長(圖4E)。相反,當[Li+]極化至0.85 M以下時,會形成富PEO相,有效抑制鋰晶須的生長(圖4F)。沉積的鋰均勻,表面[Li+]不均勻性較低。這些模擬結果與實驗結果非常吻合,證明高楊氏模量富PEO相的形成能夠抑制晶須生長。
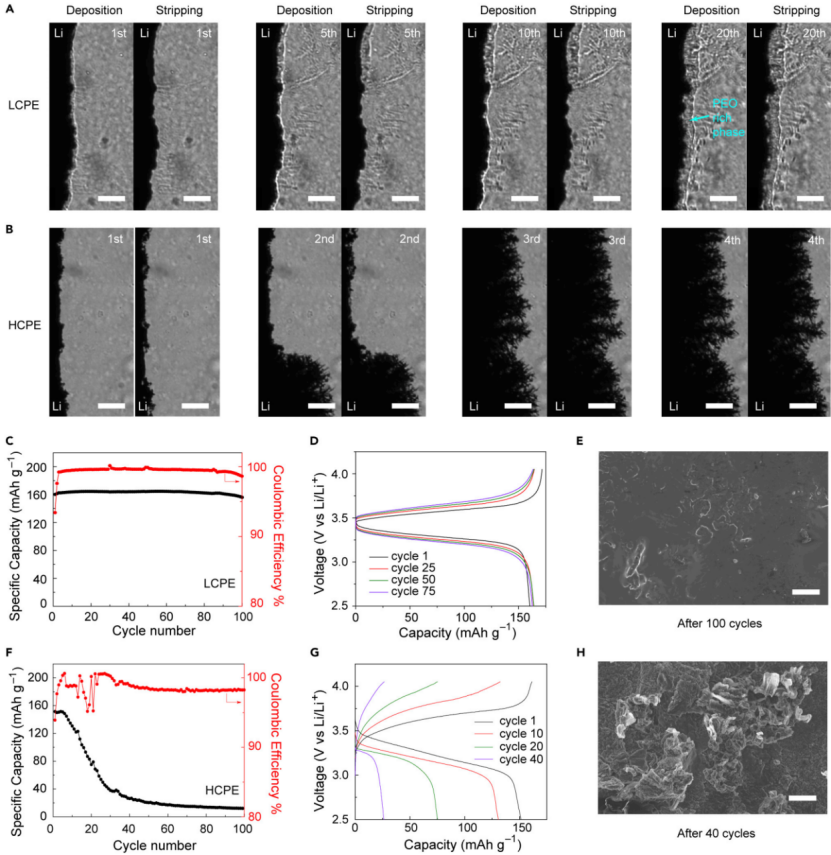
【圖5】0.5 mA cm-2下,在(A)LCPE和(B)HCPE中鋰電極電鍍和剝離的明場圖像。比例尺為100 μm。具有LCPE的LiFePEO4/Li電池(C)循環性能和(D)相應的充放電曲線。(E)循環100次后金屬鋰表面的SEM圖像。具有HCPE的LiFePO4/Li金屬電池(F)循環性能和(G)相應的充放電曲線。(H)循環40次后金屬鋰表面的SEM圖像。(E)和(H)中的比例尺為10 μm。兩個電池的電流密度為0.15 mA cm-2。所有電池在40℃下測試。
接下來,研究了這種策略在Li/Li電池和全電池循環中的有效性。首先,使Li/SN-LCPE/Li對稱電池在0.5mA cm-2@0.25 mAh cm-2下循環20次。第一次沉積時沒有形成明顯的鋰晶須,鋰突起被不斷增長的富PEO相凍結,鋰沉積穩定。在鋰剝離過程中,富PEO相和金屬鋰電極均收縮,不形成任何死鋰。20次循環后,鋰表面略有前移,表明這種抑制機制在多次循環后都是有效的。相比之下,如果沒有富PEO相,使用HCPE的Li/Li電池在前幾個循環內可以觀察到快速的晶須生長,在重復剝離過程中形成大量的死鋰(圖5B)。
圖5C顯示,使用LCPE組裝的LiFePO4/PEO/Li全電池實現了穩定的循環。初始放電容量為160.2 mAh g-1,CE為93.4%,在第10個循環中由于活化,容量緩慢增加到164.2 mAh g-1。100次循環后,容量為156.1 mAh g-1,容量保持率為97.4%。第5個循環到第100個循環的平均CE為99.5%(圖5C)。充放電曲線顯示,內阻只略有增加,沒有出現晶須引起的短路現象(圖5D)。SEM進一步顯示,100次循環后,金屬鋰表面相對平坦,偶爾出現島狀形貌,表明富PEO相抑制鋰晶須的有效性(圖5E)。
另一方面,具有HCPE的LFP/PEO/Li電池很快失效,40次循環后容量從151.5 mAh g-1下降到26.2 mAh g-1(圖5F)。充放電曲線顯示,過電位急劇增加,表明可能存在晶須生長(圖5G)。SEM圖(圖5H)證實,CE平均值僅為98.2%,這可能是由于鋰晶須生長旺盛所致。
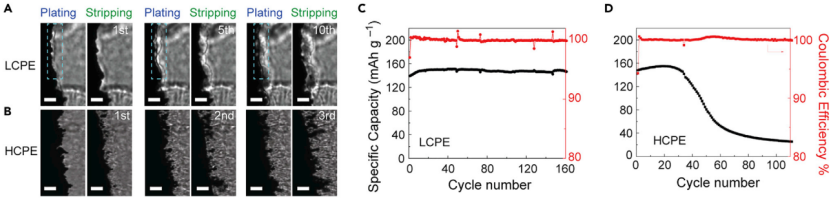
【圖6】采用PC/LiDFOB-LiBF4雙鹽基LCPE和HCPE制備的Li/Li電池和LFP/Li電池中金屬鋰負極的循環穩定性(A和B)采用(A)LCPE和(B)HCPE制備的Li/Li電池在0.75 mA cm-2@0.375 mAh cm-2下循環時,鋰負極的明場像。具有(C和D)LCPE和(D)HCPE的LiFePO4/Li金屬電池在38℃下的循環性能。電流密度為0.3 mA cm-2。在LCPE中,PEOLiBF4=121, PC為PEO的90 wt%。在HCPE中,PEOLiBF4=61, PC是PEO的90 wt%。
這種相分離誘導晶須抑制在PEO電解質中是普遍存在的。在另一種二氟硼酸鋰(LiDFOB)和四氟硼酸鋰(LiBF4)溶解在PEO/碳酸丙烯酯(PC)增塑劑的體系中,也觀察到類似的現象。首先確定了該體系中單相區和兩相區的邊界,即EO:Li+=6處。因此,在0.75 mA cm-2@0.375 mAh cm-2下對Li/PC-LCPE/Li電池進行測試,并使用光學顯微鏡對其進行表征。在鋰沉積過程中觀察到富PEO層,該層能夠抑制鋰晶須(圖6A)。相比之下,EO/Li+=3的PC-HCPE沒有出現相分離,形成了明顯的晶須和死鋰(圖6B)。圖6C顯示,LFP/PC-LCPE/Li電池在0.3 mA cm-2能夠穩定循環超過160次。而用PC-HCPE替代PC-LCPE時,100次循環后電池容量保持率僅為17.3%(圖6D)。
4、總結與展望
本工作利用具有高時間和空間分辨率的SRS顯微鏡,首次觀察到聚合物電解質中的動態濃差極化、相變以及它們與鋰沉積的相關性。結果表明,濃差極化會誘導聚合物電解質中的相變,以及電極/電解質界面上富PEO相的形成。這種相變也存在于其他各種聚合物電解質體系中。這種新相的楊氏模量高達3 GPa,通過在鋰負極上充當可逆的、自增強的保護層,有效地抑制了鋰晶須的生長。相比之下,沒有這種相變,傳統的聚合物電解質具有較小的模量《1 MPa,鋰晶須快速生長。基于上述結果,本工作提出了聚合物電解質的設計原則:在PEO-鹽-增塑劑相圖中,電解質的組成應該在單相和兩相區域的邊界處,這樣電流就可以很容易地降低金屬鋰表面的鹽濃度,從而在金屬鋰表面形成富PEO相。利用該機制,LFP/PEO/Li電池能夠穩定循環,而沒有該機制的電池在10個循環內迅速失效。該策略也與目前的電池材料制造工藝相兼容。此外,該策略對不同的鹽和增塑劑具有普適性和有效性,有利于開發具有高熱穩定性和能量密度的固體聚合物電解質基LMBs。
審核編輯 :李倩
-
電解質
+關注
關注
6文章
821瀏覽量
20652 -
固態電池
+關注
關注
10文章
726瀏覽量
28641
原文標題:哥倫比亞大學楊遠Joule:反其道而行之,濃差極化對固態電池竟是好事?
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
聚合物點焊機的原理是什么?
熱重分析儀在聚合物中的應用

清華大學:自由空間對硫化物固態電解質表面及內部裂紋處鋰沉積行為的影響
研究論文::乙烯碳酸酯助力聚合物電解質升級,提升高電壓鋰金屬電池性能

清華深研院劉思捷/港科大Kristiaan Neyts最新AEM封面文章:硫化物復合固態電解質

陳軍院士團隊最新Angew,聚合物電解質新突破

一種薄型層狀固態電解質的設計策略
半互穿網絡電解質用于高電壓鋰金屬電池
離子液體添加劑用于高壓無負極鋰金屬電池

北京科技大學范麗珍教授團隊In和F共摻雜LPSCl制備固體電解質

一種創新的超薄固體聚合物電解質
固態電池中復合鋰陽極上固體電解質界面的調控






 工商網監
工商網監
評論