 AlphaFold有望改變生物學
AlphaFold有望改變生物學
AlphaFold有望改變生物學。
近日,谷歌AI團隊DeepMind所研究的 AlphaFold 算法在生物學領域取得了重要突破:通過蛋白質的氨基酸序列高精度地確定其3D結構。
具體而言,是DeepMind的第二代AlphaFold 在國際蛋白質結構預測競賽(CASP)上擊敗了其余的參會選手,能夠精確地基于氨基酸序列,預測蛋白質的3D結構。其準確性可以與使用冷凍電子顯微鏡(CryoEM)、核磁共振或 X 射線晶體學等實驗技術解析的3D結構相媲美。
前基因泰克(Genentech)首席執行官Arthur D. Levinson博士稱這一成就為“劃時代的進步”(once in a generation advance)。
從CASP14,這項權威的蛋白質結構預測競賽結果中,可以看到AlphaFold2排名第一(總分遠高于第二),排名前六位的分別是David Barke lab的兩個工具(第二、第三位)、密歇根州立大學的 Michael Feig和密歇根大學Yang Zhang排名在第四位、第五位,而騰訊AI lab 的tFold_human 排名第六。
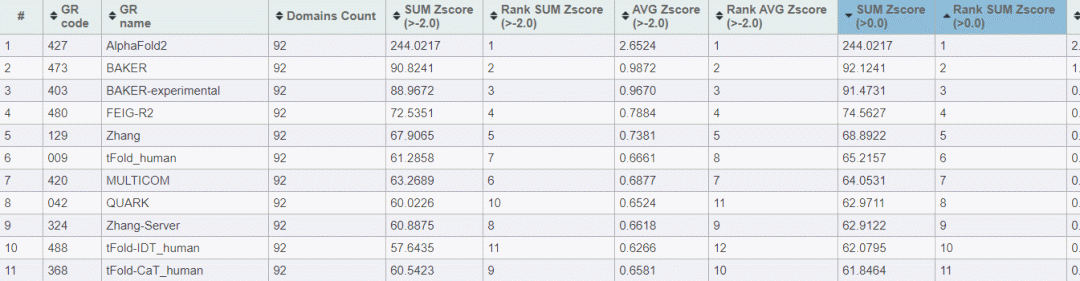
這說明,在某種程度上,在對蛋白質結構和折疊的預測這件事情上,人類已非望塵莫及。
第二代AlphaFold做了什么?
在CASP這項比賽中,DeepMind開發AlphaFold2用的數據是:數據庫里的超過17萬種蛋白序列與結構,以及其他一些大型數據庫里的數據。
評估蛋白結構預測準確度的指標叫做GDT,也就是評估預測結構里的氨基酸位置,和實際的氨基酸位置差多少。差得越少,得分越高。
GDT的分值在0-100之間。2006-2016年間,這個數字最高在40左右。2018年,上一代的 AlphaFold得分一下子突破了50。而這次的新一代 AlphaFold,在蛋白結構預測大賽里的中位得分超過了92.4。
不僅僅遠超今年的競爭對手,相比于2018年版的AlphaFold第一代,AlphaFold2的表現也如脫胎換骨一般。
更直觀一些。拿它預測的結構與實際結構對比,可以看到基本完全吻合(下圖,綠色是實驗得到的實際結構,藍色是計算預測結構)。
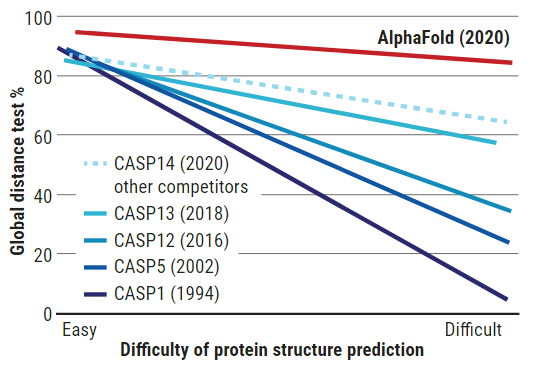
在兩年一次的蛋白質結構預測關鍵評估(CASP)競賽中,AlphaFold今年擊敗了所有其他小組,并在準確率方面與實驗結果相匹配。隨著預測難度增加,AlphaFold的準確率保持在穩定的高水平,表現遠超其它團隊以及往屆競賽的水平。
但是對此結果,也有不少人存在疑問,主要在準確性。首先,17萬的數據應該遠遠不夠,準確性存疑;再者,蛋白質結構折疊問題太過高深,如果真能夠破解,DeepMind很快就能獲得諾貝爾獎。
因此,在驚嘆AlphaFold取得成果的同時,我們還需要冷靜等待生物學家的實驗驗證。
除了解決的問題值得我們關注,其實研究方法也更有思考價值。
畢竟,AlphaFold的結構預測與X射線晶體學或低溫電子顯微鏡(cryo-EM)等標準實驗方法來預測并無區別,只是后者更加費力、更加昂貴。科學家表示,AlphaFold的出現也許并不會完全替代這些實驗方法,但確實為人們提供了研究生物的新方式。
蛋白質結構:生物學五十年來的挑戰
蛋白質是生命的基礎,與細胞組成內容緊密相關。而蛋白質的功能取決于其3D結構。
一直以來,生物學家投入實驗,致力于尋找生命的奧秘,即:氨基酸序列(蛋白質的組成部分)是如何繪制出最終的形狀。
在過去,人們一直是通過實驗室來了解蛋白質的結構。比如,使用X射線束照射結晶的蛋白質,并將衍射光轉化為蛋白質原子坐標,由此掌握蛋白質的第一個完整結構。
除了實驗,隨著計算機的發展,該項技術在上世紀末便已用于預測蛋白質的結構,但效果并不理想。
計算機技術用于蛋白質結構預測的困境,直到AlphaFold在2018年現身于CASP,才讓科學家重新燃起了信心與希望。
AlphaFold的第一次迭代將深度學習應用于結構和遺傳數據,以預測蛋白質中氨基酸對之間的距離。根據AlphaFold的主要負責人之一John Jumper介紹,在第二個步驟中,雖然沒有使用AI技術,但AlphaFold使用結構與遺傳數據得出了蛋白質的外觀模型,與之前的研究達成一致。
但第一次迭代存在缺陷。因此,該團隊開發了一個AI網絡。該網絡結合了有關確定蛋白質如何折疊的物理和幾何約束的信息。他們設立了一項艱巨的目標:該網絡能夠預測目標蛋白質序列的最終結構,而不僅是預測氨基酸之間的關系。
驚人的準確性
CASP的進行歷時數月。
1994年,Moult及其同事發起了CASP,每兩年召開一次。比賽團隊獲得約100種蛋白質的氨基酸序列,其結構未知。一些小組計算每個序列的結構,而另一些小組則通過實驗確定它。然后,組織者將計算預測結果與實驗室結果進行比較,并為預測結果提供全球距離測試(GDT)分數。
研究小組有數周的時間來提交其結構預測。然后,一組獨立的科學家使用度量標準來評估預測的蛋白質與實驗確定的結構的相似程度,以此評估各個研究小組的預測結果。研究小組的名稱為匿名。
在今年的比賽中,AlphaFold的稱號是“427組”。其預測的許多條目具有驚人的準確性,將近三分之二的預測與實驗所得的結構相當。
針對各種目標蛋白,AlphaFold的GDT中位數得分為92.4。在難度中等的蛋白質上,其他團隊的最佳表現通常在預測準確度上得分75(滿分100),而在AlphaFold則得分大約90,中位數為87,比下一個最佳預測高25分。
AlphaFold甚至擅長解決楔入細胞膜中的蛋白質結構,這是許多人類疾病的核心,但眾所周知,這個問題用X射線晶體學都很難解決。醫學研究理事會分子生物學實驗室的結構生物學家Venki Ramakrishnan稱該結果“在蛋白質折疊問題上取得了驚人的進步。”
Moult介紹,90分以上得分的預測結果與實驗方法相當。
不過,AlphaFold并非完美勝任所有的預測。在一種由52個小重復片段組成、組裝時會扭曲彼此位置的蛋白質上,AlphaFold的預測與實驗結果之間存在部分差異。
CASP的負責人Moult介紹,無法確定是因為AlphaFold的預測誤差,還是因為實驗的偽像。
此外,AlphaFold的預測與通過核磁共振成像技術確定的實驗結構的匹配度差,可能是因為AlphaFold將原始數據轉換為模型的方法需要改進。
再如,AlphaFold的網絡嘗試為蛋白質復合物或組中的單個結構建模,因此,與其他蛋白質的相互作用扭曲了它們的形狀。
應用
AlphaFold的預測有助于確定Lupas實驗室多年來試圖破解的細菌蛋白質的結構
Lupas的研究小組以前曾收集過原始的X射線衍射數據,但要將這些類似Rorschach的圖案轉換為結構,則需要了解蛋白質的結構信息。Lupas介紹:“在我們花了十年時間嘗試一切之后,427組的模型在半小時內為我們提供了結構。”
DeepMind的聯合創始人兼首席執行官Demis Hassabis介紹,AlphaFold可能需要幾天的時間才能預測出蛋白質的結構,包括對蛋白質不同區域的可靠性估計。但AlphaFold將開放給科學家使用。
Hassabis認為,AlphaFold有望應用于藥物發現和蛋白質設計。
借助AlphaFold,藥物設計人員能夠快速確定危險的新病原體(如SARS-CoV-2)中各種蛋白質的結構,這是尋找分子以阻止疾病產生的關鍵步驟。
加州大學伯克利分校的分子神經生物學家Stephen Brohawn說,DeepMind對一種叫做Orf3a的蛋白質的預測最終與后來通過cryo-EM確定的蛋白質非常相似。
AlphaFold的出現,可能意味著獲得良好的蛋白質結構,不再局限于實驗室,而只需要低質量的、易于收集的實驗數據。比方說,類似蛋白質的進化分析等方向之所以能蓬勃發展,便是因為有大量的基因組數據能夠被轉化為結構。
科學家評論,AlphaFold能夠幫助人類了解生命基因組中數千種未溶解蛋白質的功能,了解人與人之間因為疾病而產生的基因變異。
AlphaFold的出現也改寫了外界對DeepMind的印象。此前,大家知道DeepMind,一般是因為該團隊使用AI來玩游戲,比如AlphaGo。但如今,AlphaFold涉足生物學領域,例如進行蛋白質結構預測,DeepMind也因此向外界傳達了自己的另一種聲音:
不僅能玩圍棋,還能使用AI幫助生命科學的長遠發展。
責任編輯:lq
-
算法
+關注
關注
23文章
4702瀏覽量
94990 -
數據庫
+關注
關注
7文章
3905瀏覽量
65899 -
DeepMind
+關注
關注
0文章
131瀏覽量
11439
原文標題:劃時代的進步!DeepMind第二代AlphaFold破解生物學“五十年難題”,可精準預測蛋白質3D結構
文章出處:【微信號:IoT_talk,微信公眾號:醫健AI掘金志】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
仿生傳感器:讓機器擁有“生命感知”的神奇科技
生物傳感器:解鎖生命密碼的“芯片”,正在改變世界的感知方式
生物傳感器的主要組成包括_生物傳感器的分類
SSTF中脈沖前傾的仿真
淺談生物傳感技術的定義、發展現狀與未來
恒溫箱內的敏銳氣體偵探-氧化鋯氧傳感器SO-E2-250、SO-E2-960

微流控技術的生物學應用
生物傳感器:科技前沿的生物監測利器
NVIDIA 推出 BioNeMo 開源框架,擴大全球生物制藥和科學行業的數字生物學研究規模
氧化鋯氧傳感器SO-E2-XXX在恒溫箱內的應用






 工商網監
工商網監
評論