 一種具有自發電子存取能力的新型電子存儲器
一種具有自發電子存取能力的新型電子存儲器

一、 全文概要
由于Li?S電池存在復雜的多電子、多相SRR反應,通常會導致電荷轉移動力學緩慢,以及電池內可溶性LiPSs的積累和穿梭。引入電催化劑加速LiPSs轉化,被證實是解決上述瓶頸的有效方法。稀土元素由于其f軌道部分填充的電子構型,被認為是很有前途的電子存儲器(電子可存取)。同時,我們注意到稀土氧化物具有豐富的電子結構、可變的價態和配位數,可用于同步催化復雜多電子反應體系。在17種稀土元素中,鋱(Tb)元素具有價態豐富、氧化性強、無放射性的特點。通常情況下,4f層有8個電子的Tb為+3價,記為Tb3+,它可以作為電子源向外界提供電子(取電子);+4價的Tb,記為Tb4+,含有半填充的4f殼層,其具有高氧化能力和3.3 V vs. NHE的高氧化還原電位,可以作為充電過程中硫氧化反應的潛在氧化劑,賦予Tb存儲電子的能力(存電子)。受此啟發,基于鋰硫電池體系,我們利用Tb3+/Tb4+復合氧化物開發了一種具有自發電子存取能力的新型電子存儲器。

溫州大學楊植教授、楊碩博士與蔡冬博士聯合設計開發了具有電子適中填充f軌道的Tb3+/4+氧化物作為電子存儲器用于鋰硫電池。通過一系列原位/非原位表征技術以及DFT理論計算分析表明,Tb電子存儲器能夠在Li?S電池充放電過程中動態釋放/接收電子,并通過Tb?S和N???Li鍵吸附LiPSs,降低活化能壘,加速電子和Li+傳輸,在充放電過程中選擇性催化長鏈和短鏈LiPSs轉化。引入Tb3+/4+電子存儲器的Li?S電池在高硫載量和貧電解液條件下(面密度為5.2 mg cm?2,液/硫比為7.5 mL mg?1)展現了穩定的循環性能(1 C循環500次,單圈衰減率為0.087%)。相關研究成果以“Regulating f orbital of Tb electronic reservoir to activate stepwise and dual-directional sulfur conversion reaction”為題發表在《InfoMat》(IF=24.789)雜志上。
二、 研究亮點
1、通過解耦每一步硫轉化所需的催化劑條件,合理設計了具有適中f軌道的混合價Tb(Tb3+/4+)作為電子存儲器來降低每一步硫轉化的活化能壘,加速界面電子/Li+傳輸動力學。
2、通過多種表征方法證實了,Tb3+促進長鏈多硫化鋰的轉化,Tb4+促進短鏈多硫化鋰的轉化,并通過重要的中間體S52?,完成了充放電過程中不同鏈長LiPSs之間的轉化反應。
3、本研究論文展現了f軌道調控策略在開發高性能Li?S電池體系方面的潛力。
三、正文導讀
1、DFT計算
催化劑材料的本征電子結構決定了它們與LiPSs的結合強度和催化硫轉化的能力。為了預測不同價態Tb氧化物吸附催化LiPSs的本質情況,首先利用DFT計算了不同價態Tb氧化物的電子結構(圖1)。考慮到Tb氧化物催化劑在SRR過程中的非活性聚集和催化劑體系的導電性,在催化劑的模型構建中引入了N摻雜的石墨烯(將其標記為Gh)以更好的錨定催化劑,改善體系的電子傳輸。其中4個Tb原子與不同數量的O原子配位得到3種Tb價態(Tb3+、Tb3+/4+和Tb4+分別配位6、7和8個O原子)。Gh-Tb3+的f帶中心相對較高,根據d帶電子理論說明其向外電路提供電子的能力和對外界物質的吸附能力更強,形成Tb3+電子源。同樣,Gh-Tb4+的f帶中心最低,更容易接受來自外界的電子,對外界物質表現出弱吸附能力,形成Tb4+電子漏。相比之下,Gh-Tb3+/4+在三種復合材料中表現出中等的f帶中心,表明其對被吸附物質(如LiPSs)的吸附能力適中,增益和損失電子的能力平衡(即產生電子存儲器(源和漏))。f帶中心的結果得到了材料與LiPSs之間結合能計算的有力支持。LiPSs分子可通過Tb?S鍵吸附在Gh-Tb3+/4+表面,與其他三種材料(Gh-Tb3+,Gh-Tb4+,Gh)相比,Gh-Tb3+/4+對大部分的LiPSs(Li2Sn,n = 8、4、1)分子都表現出更適中的吸附強度。這意味著Gh-Tb3+/4+作為電子存儲器不僅能有效錨定LiPSs,而且能使LiPSs的后續轉化達到理想的水平。S8還原到Li2S8的過程的吉布斯自由能壘顯示在所有基底上都是自發的放熱反應。從Li2S8到Li2S4的兩個長鏈LiPSs轉化過程,在Gh-Tb3+是為放熱反應,較容易進行;雖然在Gh-Tb3+/4+基底上是吸熱反應,但是Gh-Tb3+/4+上的吉布斯自由能壘相對于Gh-Tb4+和Gh較小。而對于形成短鏈Li2S的吉布斯自由能勢壘,含Tb4+的基底遠低于Gh-Tb3+和Gh。因此,可以確定Tb3+和Tb4+分別能有效降低長鏈LiPSs轉化反應和短鏈LiPSs轉化反應的吉布斯自由能壘。重要的是在Gh-Tb3+/4+基底上各個步驟的吉布斯自由能壘普遍較低,特別是在Li2S6到Li2S4這一步,最低值為0.12 eV,因此在放電過程中,從S8到Li2S的整個硫轉化過程在Gh-Tb3+/4+表面上更加容易進行。從電子轉移的角度分析,我們推斷Tb3+電子源可自發地向長鏈LiPSs提供電子,牢牢地捕獲長鏈LiPSs并迅速地將其轉化為短鏈LiPSs;隨后在放電過程的推動下,作為存儲電子的Tb4+電子漏以吸附弱,強催化的原則繼續將短鏈LiPSs進一步轉化為放電產物Li2S,保證硫還原過程的連續性。借助于Tb3+和Tb4+的共存,在Tb3+/4+電子存儲器(電子源和電子漏)上的電子轉移和電化學反應能夠更加高效進行。DFT計算研究認為,Gh-Tb3+/4+由于具有中等的f帶中心和電子得失能力,并且可以通過Tb?S鍵以合適的作用力捕獲LiPSs,而不會影響LiPSs后續催化轉化過程,有望提高Li?S電池的電化學性能。
2、Tb電子存儲器的制備與表征
我們制備了三種Gh-Tb氧化物(Gh-Tb3+、Gh-Tb3+/4+和Gh-Tb4+),并對其結構進行表征(圖1)。在空氣條件下熱處理制備的Gh-Tbx+復合材料的Tb 3d XPS數據表明形成了混合價態Tb,即形成Gh-Tb3+/4+。N 1s XPS光譜中存在吡啶/吡咯N峰,表明Tb分子中的N元素熱分解后形成了N摻雜Gh。為了控制Tb價態為單一變量,在接下來的研究中,我們將采用N摻雜的Gh制備物理混合的Gh-Tb3+和Gh參比樣品。Gh-Tb3+/4+的TEM圖可以看到,充分均勻分布的Tb氧化物顆粒覆蓋了Gh的整個表面。在Gh-Tb3+/4+復合物的HAADF-STEM圖像發現Tb氧化物以超微團簇的形式存在。Gh-Tb3+/4+的EDX元素映射圖進一步證實了C、N、O和Tb元素在結構中均質共存,與XPS結果一致。N摻雜Gh的高電導率和大活性比表面積特性,以及結合Tb3+/4+獨特的電子結構在LiPSs吸附和催化方面的優異潛力,因此,認為Gh-Tb3+/4+能夠應用于Li?S電池正極改性工作中。將Gh-Tb3+/4+作為改性正極的插層膜結構以促進SRR動力學,緩解Li?S電池的穿梭效應,來提高Li?S電池的性能。
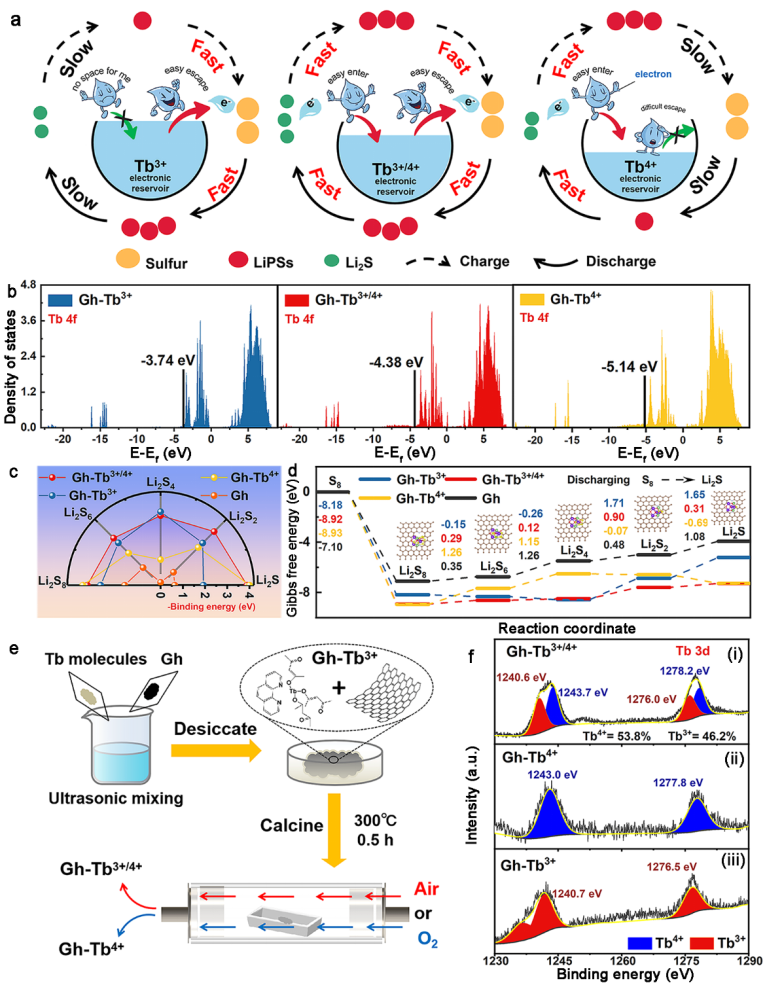
圖1催化劑的設計、理論模擬和表征及其與LiPSs的相互作用(a)不同Tb電子存儲器SRR機理示意圖。(b)Gh-Tbx+(x = 3, 4, 3+/4)的Tb 4f軌道的態密度。(c)Li2Sn(n = 8,6,4,2,1)與Gh-Tb3+、Gh-Tb3+/4+、Gh-Tb4+和Gh基底之間的結合能。(d)四種催化劑表面硫還原過程的吉布斯自由能變化。(e)Gh-Tbx+(x = 3, 4, 3+/4)復合材料的合成過程示意圖。(f)Gh-Tbx+(x = 3, 4, 3+/4)的Tb 3d XPS光譜。
3、Tb電子存儲器對LiPSs的化學吸附和轉化動力學研究
盡管Gh-Tb3+/4+的比表面積和中孔體積都小于Gh,但由于Gh-Tb3+/4+對LiPSs的物理和化學協同作用,加入Gh-Tb3+/4+樣品的Li2S6溶液的顏色幾乎變為無色,這也更加證實了Gh-Tb3+/4+具有優異的LiPSs吸附能力(圖2)。進一步分析Gh-Tb3+/4+吸附Li2S6溶液前后的XPS數據,在Tb 3d XPS譜中,所有Tb 3d峰在吸附Li2S6溶液后明顯向更高的結合能方向移動,表明Tb原子周圍的電子云密度降低。樣品吸附Li2S6溶液后的S 2p XPS譜中,Gh-Tb3+/4+復合材料在164.1/165.2 eV處S?S鍵的峰增強,此外在162.6/163.7 eV處出現了一對額外的新峰歸屬于硫和金屬形成的化學鍵(S?M)。以上結果表明,Gh-Tb3+/4+與LiPSs之間通過Tb?S鍵保持著良好的化學親和性。同時,在Li 1s譜54.5 eV處出現了更強的Li???N的峰,表明N摻雜的Gh與LiPSs之間形成了類似“鋰鍵”的結構,在DFT吸附優化模型中彎曲Li2Sn分子上的Li原子與Gh的N原子相鄰,也支持了鋰鍵的形成。以上結果再次證實了Gh-Tb3+/4+與LiPSs之間存在良好的化學相互作用力。
通過對稱電池測試技術評估了四種樣品對可溶性LiPSs的液-液轉化過程的動力學行為。CNTs/ Gh-Tb3+/4+對稱電池表現出最小的反應極化、最高的氧化還原電流響應和最明顯的氧化還原峰。此外,對四種材料電極的對稱電池進行EIS測試也得到了類似的結果。含有Tb3+的對稱電池的Rct明顯小于其他兩種電池的Rct,特別是CNTs/Gh-Tb3+/4+對稱電池的Rct最低,表明其電荷轉移最快,進而促進了SRR。恒電位條件下測試的Li2S沉積/分解實驗為分析研究液固轉化動力學提供了重要的參考依據。整體來說CNTs/Gh-Tb3+/4+電極與其他三種電極相比,展現出最快的Li2S成核響應速率(3604 s),最大的響應電流密度(0.45 mA)和最高的Li2S沉積比容量(351 mAh g?1),表明在CNTs/Gh-Tb3+/4+表面加快了Li2S沉積動力學。CNTs/Gh-Tb3+/4+電極在Li2S分解實驗中顯示出與Li2S成核相似的實驗現象。值得注意的是CNTs-S/Gh-Tb4+正極在Li2S沉積和分解過程中表現出第二好的性能,這意味著Tb4+能夠很好地促進短鏈LiPSs轉化(液-固/固-液轉換)。由GITT計算的DLi+結果顯示,Tb3+/4+的DLi+相比于其他的電池提升了一個量級,特別是從液態Li2S4到固態Li2S2/Li2S,證實了CNTs-S/Gh-Tb3+/4+在促進Li+擴散和激活鋰化動力學方面的優異能力。Ea可以從根本上直接反映出上述涉及液-液和液-固轉化的SRR反應動力學,整個放電過程在長鏈LiPSs轉化的電壓范圍內(2.8~2.1 V),Tb3+樣品的Ea低于Tb4+樣品,而在短鏈LiPSs轉化的電壓范圍內Tb4+樣品的Ea較低。充電過程也呈現了相似的結果。以上結果再次表明,Tb3+和Tb4+可以分別促進長鏈和短鏈LiPSs轉化。此外,由于Tb3+/4+可以降低硫轉化過程中的能量勢壘,顯著加快硫雙向轉化動力學同時提高硫的利用率,含有Tb3+/4+的電池在充放電過程的每一步活化能Ea值都是相對低的,這有望提高電池性能。
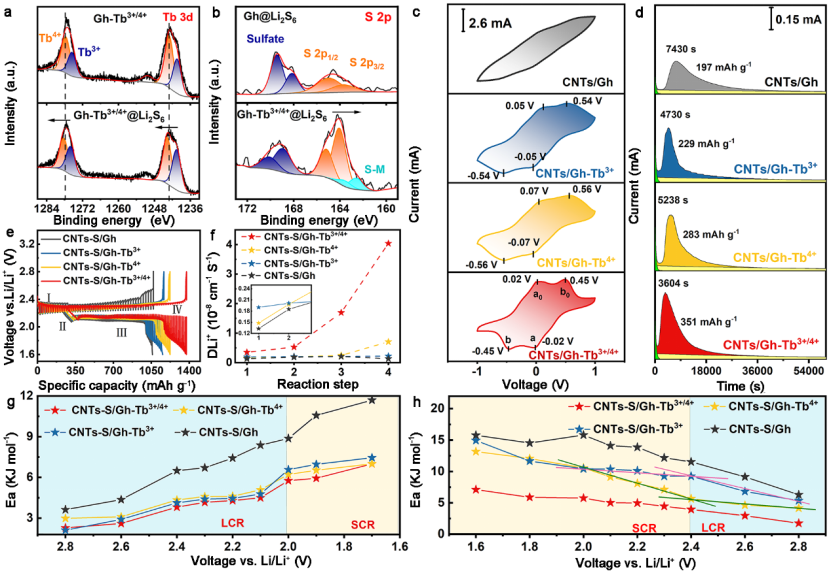
圖2多相SRR的吸附能力和動力學評價(a)Gh-Tb3+/4+吸附Li2S6前后的高分辨Tb 3d XPS光譜。(b)Gh和Gh-Tb3+/4+與Li2S6相互作用后的高分辨S 2p XPS譜。(c)以Li2S6電解液為活性組分,CNTs/Gh-Tbx+(x = 3, 4, 3+/4)和CNTs/Gh為電極的對稱電池在30 mV s?1時的CV曲線。(b)在CNTs/Gh-Tbx+(x = 3, 4, 3+/4)和CNTs/Gh正極上沉積Li2S的恒壓放電曲線。 綠色和淺黃色區域分別代表Li2S8和Li2S6的還原。(e)CNTs-S/Gh-Tbx+(x = 3,4,3+/4)和CNTs-S/Gh正極(電流脈沖:83.7 mA g?1,脈沖10 min,弛豫2 h)第二周期的GITT充放電曲線。(f)四種電極在放電過程中的DLi+。(g)在放電和(h)充電過程中四種電極SRR過程中活化能的分布。
4、Tb電子存儲器在鋰硫電池中的獨特作用探究
采用半原位XPS研究了不同催化劑的作用機理(圖3)。根據充放電過程中不同硫物種的峰強度演化,可以分析不同催化劑對硫的催化轉化能力。在充滿電的狀態下(2.8 V)所有正極的表面成分均以S8為主。有趣的是,當電池放電到2.0 V時,在含有Tb3+的正極S8信號顯著減弱,長鏈LiPSs(Li2Sn, 4 ≤ n ≤ 8)的信號顯著增強,說明Tb3+對S8向長鏈LiPSs的轉化具有良好的催化作用。放電結束時(1.6 V),含有Tb4+的正極比無Tb4+的正極(CNTs-S/Gh-Tb3+和CNTs-S/Gh)顯示出更強的Li2S2/Li2S信號,此外含有Tb4+的正極仍存在部分長鏈LiPSs(Li2Sn,4 ≤ n ≤ 8)信號,說明Tb4+在促進長鏈LiPSs之間轉化的能力是有限的,但是可以有效的催化短鏈LiPSs向Li2S2/Li2S的轉化,這種現象在充電過程中也是高度可逆的。由于Tb3+和Tb4+特殊的電子結構以及協同催化作用,CNTs-S/Gh-Tb3+/4+正極在各充/放電狀態下表現出最佳的硫化物信號,體現出連續催化LiPSs轉化的非凡能力。
電極/電解液界面的原位UV-vis光譜及其相應的吸光強度的定性分析進一步支持了這一說法(圖3),與不含Tb3+的正極相比,含Tb3+的正極在放電過程中S82?/S42?強度下降更快,同時S3*?信號相應的增強,再次證實Tb3+更傾向于加速長鏈LiPSs向短鏈LiPSs的轉化。充電過程中CNTs-S/Gh-Tb3+正極在~2.3~2.8 V電壓區間,S42?/S62?信號的變化進一步證明了以上闡述的觀點。值得注意的是,在放電時CNTs-S/Gh-Tb4+正極S3*?的吸光強度不斷下降,同時在充電時含有Tb4+的正極,S32?的吸光強度在快速衰減,S42?/S62?的吸光強度隨著充電的進行在不斷增加,這同樣表明Tb4+可以促進短鏈LiPSs的轉化。因此,CNTs-S/Gh-Tb3+/4+在放電/充電過程中,能夠有效促進長鏈和短鏈LiPSs的最佳轉化,歸結于Tb3+和Tb4+電子結構耦合的效果。
通過原位拉曼實時監測LiPSs在不同含硫正極上的轉化途徑(圖4)。在初始放電狀態下(2.8 V),在CNTs-S/Gh正極表面,在154、219和473 cm?1波數處有三個明顯的S8峰,完全放電到1.6 V時,S8的強度減弱。然而,由于CNTs-S/Gh的催化能力有限,在整個過程中沒有出現短鏈LiPSs的信號。對于CNTs-S/Gh-Tb3+和CNTs-S/Gh-Tb4+正極的原位拉曼光譜顯示了和CNTs-S/Gh類似的S8轉化;除此之外,CNTs-S/Gh-Tb3+正極放電到約2.2 V時在120和495 cm?1處出現了兩個弱峰對應S52?和S32?+ S42?信號;CNTs-S/Gh-Tb4+正極放電到2.1~2.0 V時在447 cm?1處出現了S42?+Li2S2信號,再次體現了Tb3+和Tb4+分別在促進長鏈和短鏈LiPSs轉化中的作用。對于CNTs-S/Gh-Tb3+/4+正極,隨著放電的進行除了S8信號逐漸減弱之外,在放電到中間態(2.3~2.2 V)時出現了明顯的S62?和S52?的信號,并且在后續的放電過程中隨著S62?和S52?信號的減弱,出現了Li2S2的信號并在逐漸增強,以上結果表明在CNTs-S/Gh-Tb3+/4+正極上,S8通過S62?/S52?/S42?中間產物徹底的轉化為放電產物Li2S/Li2S2。所以在Li2S6向Li2S4轉化階段的低吉布斯自由能壘是由于新的Li2S5中間體的形成,在Li2S6向Li2S4轉化時起到了跳板的作用。
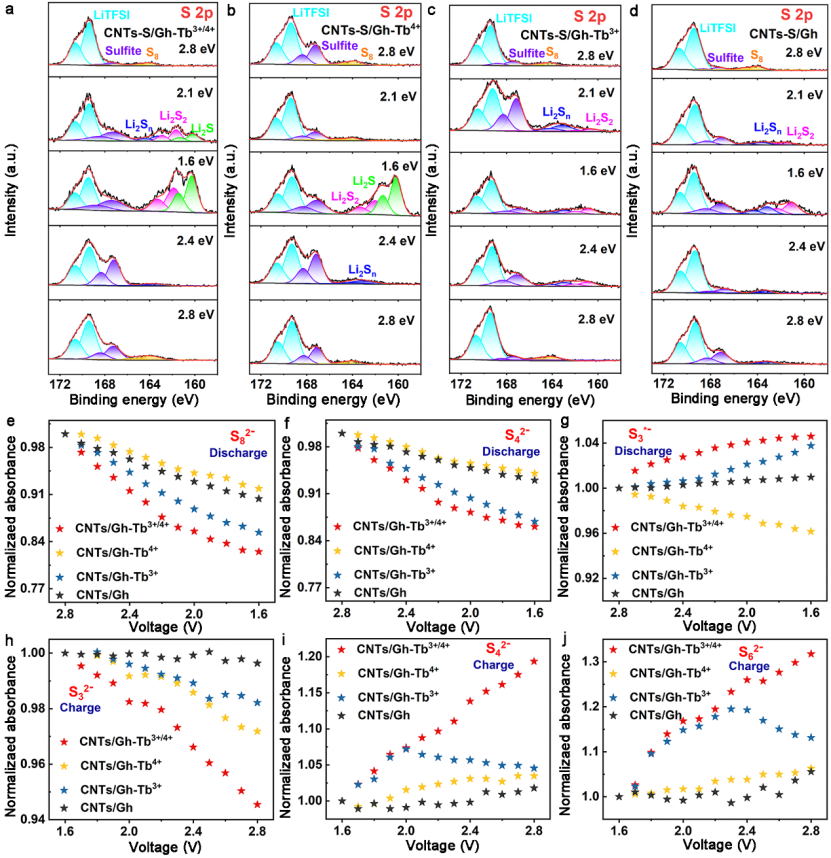
圖3 Tb電子存儲器對LiPSs的催化作用(a)CNTs-S/Gh-Tb3+/4+(b)CNTs-S/Gh-Tb4+(c)CNTs-S/Gh-Tb3+和(d)CNTs-S/Gh電極在放電和充電到特定電位后的半原位S 2p XPS光譜。(e-g)放電和(h-j)充電時CNTs-S/Gh-Tb3+/4+、CNTs-S/Gh-Tb4+、CNTs-S/Gh-Tb3+和 CNTs-S/Gh在Li2S8/Li2S3溶液中的原位紫外可見吸收光譜歸一化處理。
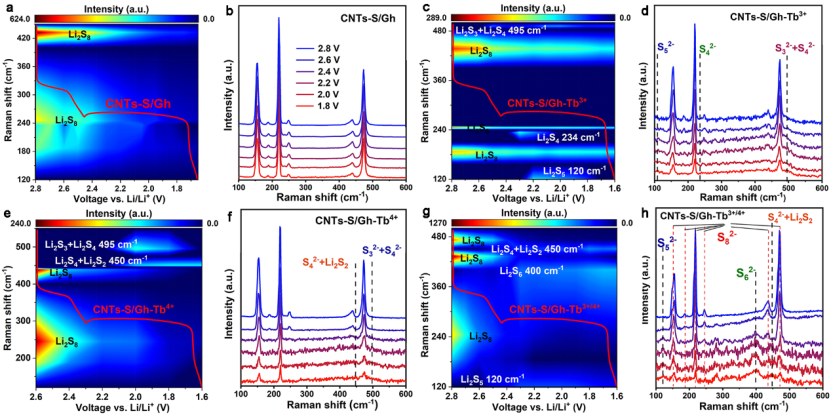
圖4 LiPSs在不同正極上的轉化途徑。放電過程中(a,b)CNTs-S/Gh-Tb3+/4+,(c,d)CNTs-S/Gh-Tb4+,(e,f)CNTs-S/Gh-Tb3+和(g,h)CNTs-S/Gh電極的原位拉曼光譜和相應的強度變化曲線。
5、Tb電子存儲器在鋰硫電池中的性能評估
CNTs-S/Gh-Tb3+/4+電池的CV曲線經初始活化后在后幾圈的循環中幾乎重疊,這意味著電池反應具有良好的可逆性。與CNTs-S/Gh-Tb4+、CNTs-S/Gh-Tb3+和CNTs-S/Gh電池相比,CNTs-S/Gh-Tb3+/4+電池的第二個還原峰和第一個氧化峰之間的分離最小,同時也表現出最正的還原起始電位和最負的氧化起始電位,說明加入Tb3+/4+后的電池對硫轉化的能量屏障最低,對氧化還原反應的抵抗力最低,具有最小的電化學極化和最佳的電化學動力學行為(圖5)。與其他電池相比CNTs-S/Gh-Tb3+/4+電池的電荷轉移電阻(Rct)和電解液電阻(Re)最低。在電池充放電期間,Re的最大值對應于電解液中LiPSs的濃度峰值,隨后Re的降低與LiPSs進一步轉化導致其濃度降低有關。相比之下,含有Tb3+/4+電池的Re是最小的,這反映了Tb3+/4+能夠有效錨定LiPSs并催化其轉化。充電過程中也會出現同樣的現象,并且Re峰值開始下降的電位提前,這進一步證明反應動力學得到了改善。根據CV曲線中的還原峰和氧化峰計算了Tafel斜率,Tb3+/4+的加將P1、P2和P3處的Tafel斜率降到最低值,表明選擇性催化策略分別賦予了SRR過程42~60%的動力學增強。對0.2 C下的第二圈的充放電平臺曲線進行分析,CNTs-S/Gh-Tb3+/4+電池的電壓極化最小為18 mV,CNTs-S/Gh-Tb3+/4+在~2.3 V處的第一個平臺容量(Qup)值為392 mAh g?1接近理論容量(418 mAh g?1),與其他三個電池相比第二個平臺(Qlow)延長了24%,這表明CNTs-S/Gh-Tb3+/4+對活性物質硫的利用率更高,液-固轉換進行的更快,再次說明了Gh-Tb3+/4+的特殊催化作用,加速了Li2S沉積和液-固轉化動力學。
CNTs-S/Gh-Tb3+/4+電池在硫負載量約為1.0 mg cm?2的情況下可以實現良好的長循環性能,1 C倍率下的初始放電比容量超過1157 mAh g?1,硫利用率高達70%,在循環500次后比容量可以穩定地維持在656 mAh g?1以上,單圈容量衰減率僅為0.087%,循環穩定性得到了顯著的改善,并且在整個循環過程中的平均庫侖效率高于99%,說明具有較高的硫利用率和成功捕獲可溶性LiPSs的能力。此外,CNT-S/Gh-Tb3+/4+在0.5 C下的長循環測試中提供了約1115 mAh g?1的初始放電比容量,并在連續108次循環后放電比容量保持在990 mAh g?1,容量保持率高達89%。
硫的高負載能力是擴大Li?S電池實際應用的關鍵基礎,因此我們進一步將面積硫載量增加到4.1 mg cm?2,在0.1 C下的初始放電比容量為993 mAh g?1循環120圈后放電比容量為765 mAh g?1,而CNTs-S/Gh-Tb4+、CNTs-S/Gh-Tb3+和CNTs-S/Gh正極的電池放電比容量顯著衰減,無法穩定循環。這一比較進一步證明了在高載硫電極中Tb3+和Tb4+的同時參與,作為電子存儲器加速放電和充電過程的重要性。后又進一步在嚴苛的條件下對使用Gh-Tb3+/4+催化劑電池的實用性進行評估(電極面密度為5.2 mg cm?2,E/S比為7.5 mL mg?1)。在0.05 C放電倍率下Gh-Tb3+/4+催化劑仍能保證貧電解液情況下的Li?S電池具有高達1225 mAh g?1的初始放電比容量,在經歷134圈循環后容量衰減率為0.18%。
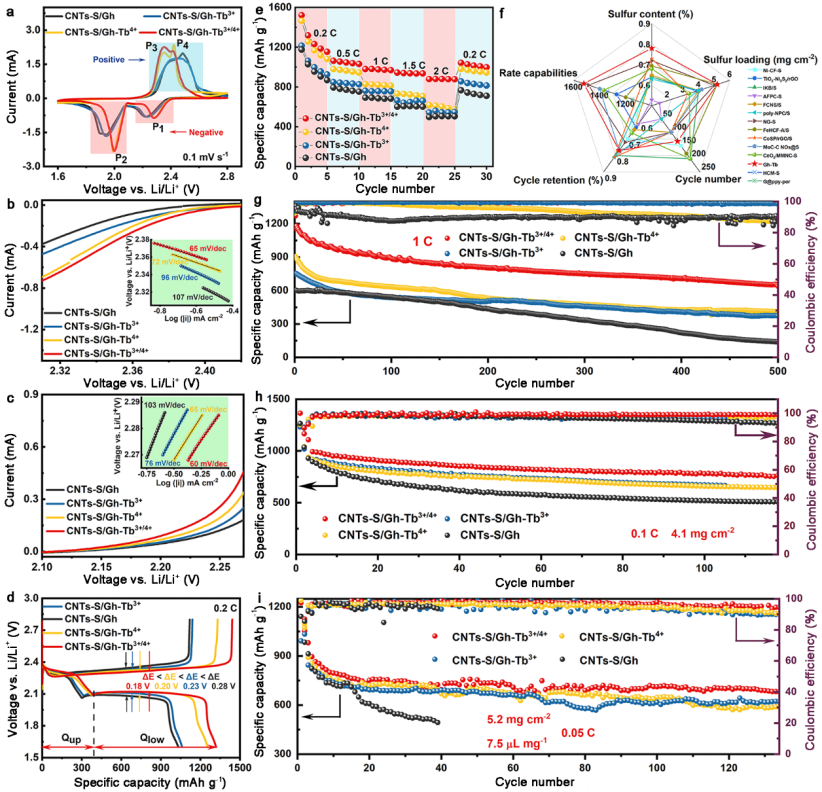
圖5 Li?S電池電化學性能(a)CNTs-S/Gh-Tb3+/4+、CNTs-S/Gh-Tb4+、CNTs-S/Gh-Tb3+和CNTs-S/Gh正極在0.1 mV s?1下的CV曲線,電位介于1.6和2.8 V之間。從圖5(a)中的CVs獲得的還原和氧化過程的Tafel斜率,對應于(b)P1和(c)P3。(d)CNTs-S/Gh-Tb3+/4+、CNTs-S/Gh-Tb4+、CNTs-S/Gh-Tb3+和CNTs-S/Gh正極在0.2 C的恒電流放電/充電平臺曲線。(e)CNTs-S/Gh-Tb3+/4+、CNTs-S/Gh-Tb4+、CNTs-S/Gh-Tb3+和CNTs-S/Gh正極0.2 C~2 C之間的倍率性能。(f)CNTs-S/Gh-Tb3+/4+電極與其他電極體系的性能比較。(g)四種電極在1 C下的長循環性能(含硫量:1.0 mg cm?1)。(h)0.1 C下的長期循環性能(含硫量:4.1 mg cm?2)。(i)經0.02 C循環三圈活化后在0.05 C下的長期循環性能(含硫量:5.2 mg cm?2,E/S比為7.5 mL mg?1)。
四、總結與展望
本研究提出了通過調節Tb電子存儲器f軌道的策略,來實現平穩、連續的硫轉化。通過解耦每一步硫轉化所需的催化劑條件,合理設計了具有適中電子填充f軌道的Tb3+/4+作為電子存儲器催化劑應用到Li?S電池中來降低每一步硫轉化的活化能壘,加速界面電子/Li+傳輸動力學,并通過一個重要的中間體S52?完成了充放電過程中不同鏈長LiPSs之間的轉化反應。憑借Tb3+/4+獨特的電子結構,CNTs-S/Gh-Tb3+/4+電池在0.2 C下的初始放電比容量高達1522 mAh g?1,在1 C下能夠穩定循環500圈,容量衰減率僅為0.087%,顯示出較好的循環穩定性。此外,在硫面密度為5.2 mg cm?2、E/S比為7.5 mL mg?1的情況下也能夠正常工作,展示了f軌道調控策略在開發高性能Li?S電池體系方面的潛力。
審核編輯 :李倩
-
存儲器
+關注
關注
38文章
7644瀏覽量
166913 -
電荷
+關注
關注
1文章
652瀏覽量
36717 -
催化劑
+關注
關注
0文章
93瀏覽量
10515
原文標題:楊植InfoMat:稀土元素f軌道調控鋰硫電池中硫的可逆催化
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
閃速存儲器屬于RAM還是ROM,閃速存儲器一般用來做什么的
內存儲器的分類和特點是什么
隨機內存儲器的特點有哪些
DRAM存儲器的特性有哪些
靜態隨機存儲器的定義和工作原理





 工商網監
工商網監
評論