 使用AlphaFold2進行蛋白質結構預測
使用AlphaFold2進行蛋白質結構預測

前言
AlphaFold 2,是DeepMind公司的一個人工智能程序。2020年11月30日,該人工智能程序在蛋白質結構預測大賽CASP 14中,對大部分蛋白質結構的預測與真實結構只差一個原子的寬度,達到了人類利用冷凍電子顯微鏡等復雜儀器觀察預測的水平,這是蛋白質結構預測史無前例的巨大進步。這一重大成果雖然沒有引起媒體和廣大民眾的關注,但生物領域的科學家反應強烈。
目前,AlphaFold2的源代碼已經在GitHub上公開,而且現在科學家正在利用AlphaFold2對已有的蛋白數據庫進行高通量的預測,建立了一些模式生物物種所有蛋白的AlphaFold2預測結構數據庫。
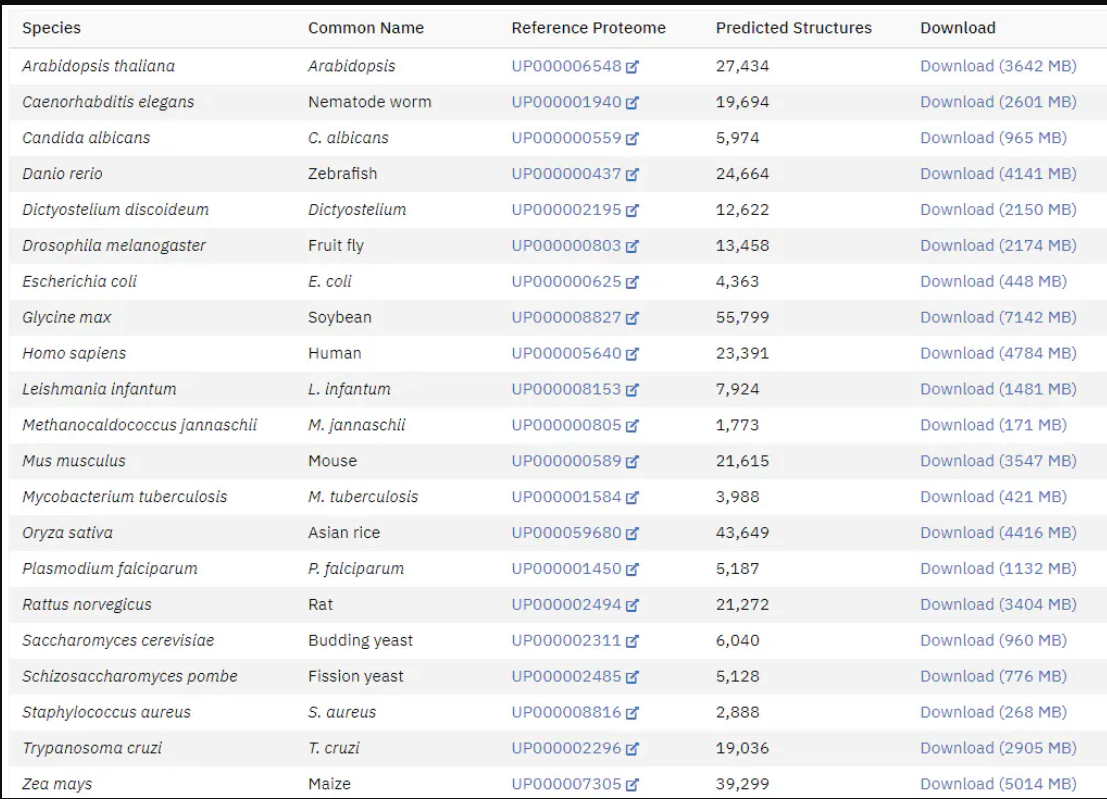
可以看到,雖然利用AlphaFold2預測了這么多生物的數據庫,但是并未覆蓋所有的蛋白序列數據庫,所以只有搭建本地的AlphaFold2服務,你才能用AlphaFold2隨心所欲的預測自己研究蛋白的結構。
接下來將給大家介紹AlphaFold2的使用方法,在北鯤云上免安裝使用。對于沒有Linux基礎或本地硬件配置不足的人,僅需1分鐘即可成功提交蛋白質結構預測任務,能夠省去很多麻煩。
二、在北鯤云使用AlphaFold2進行蛋白質結構預測
1. 選擇AlphaFold2
在“應用中心”搜索AlphaFold2軟件并選中,在右側彈出的軟件詳情欄中點擊“提交作業”。
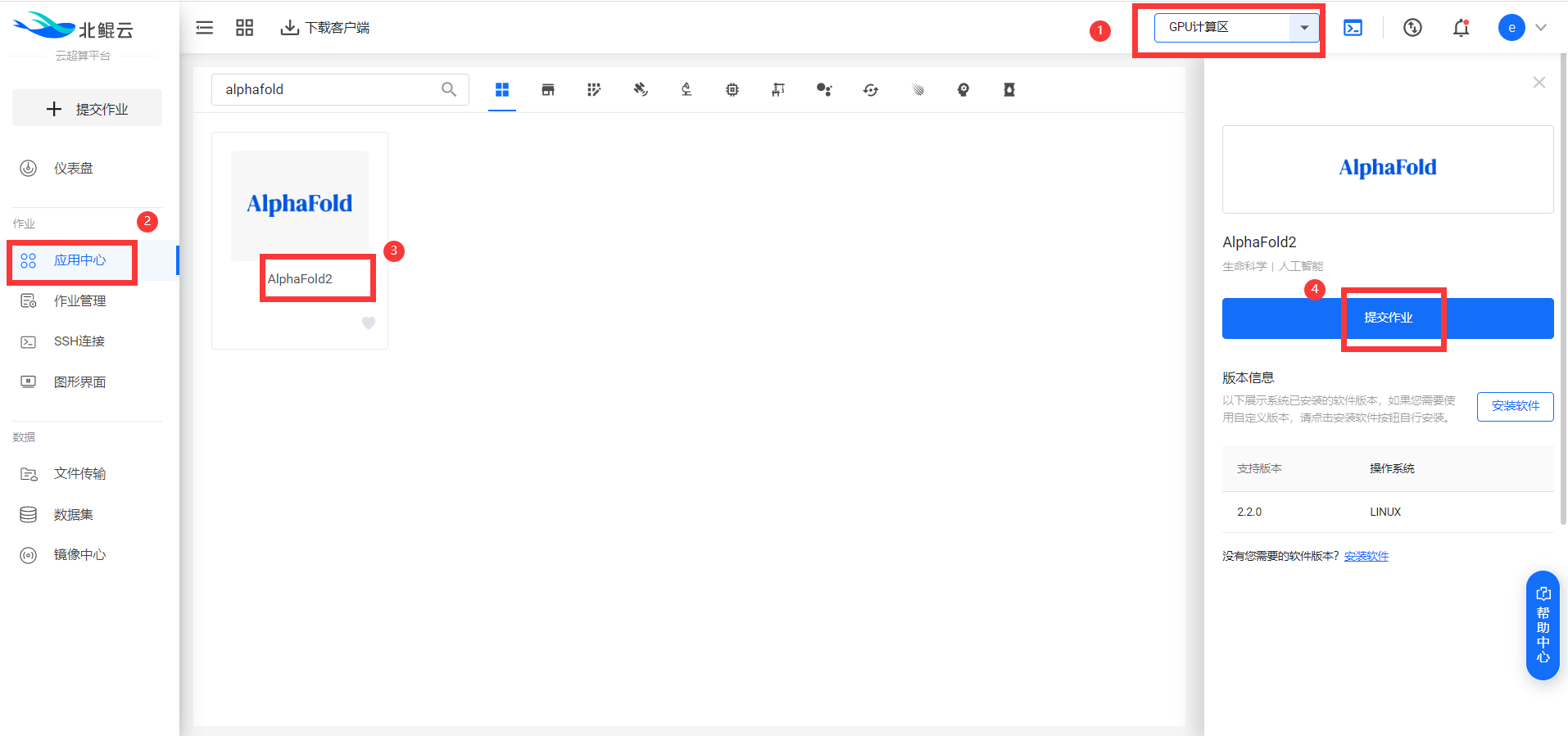
2. 選擇可視化模板提交
推薦選擇可視化“模板提交”的方式提交作業,平臺已為AlphaFold2內置了幾個可視化模板,按要求填寫相應參數即可提交預測任務。
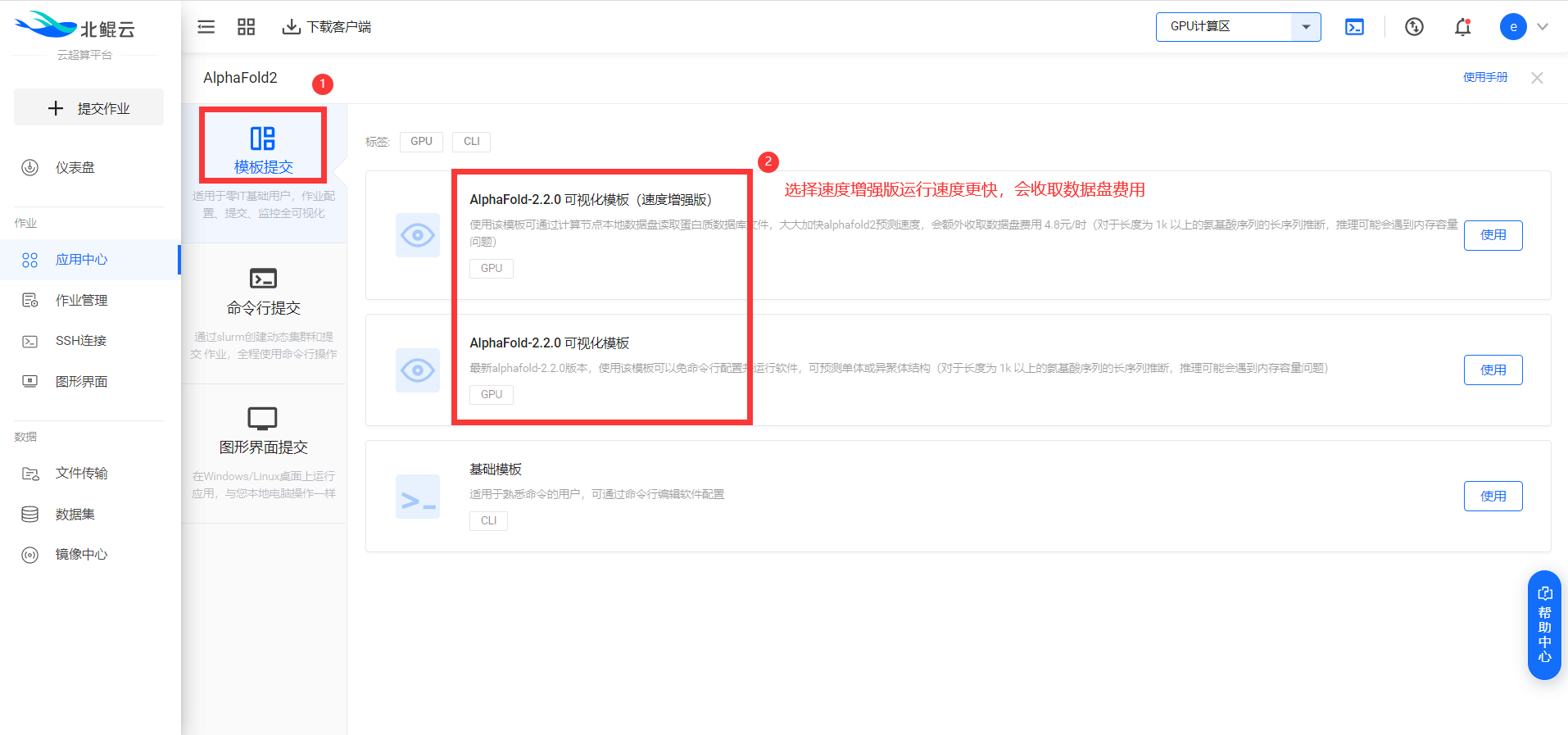
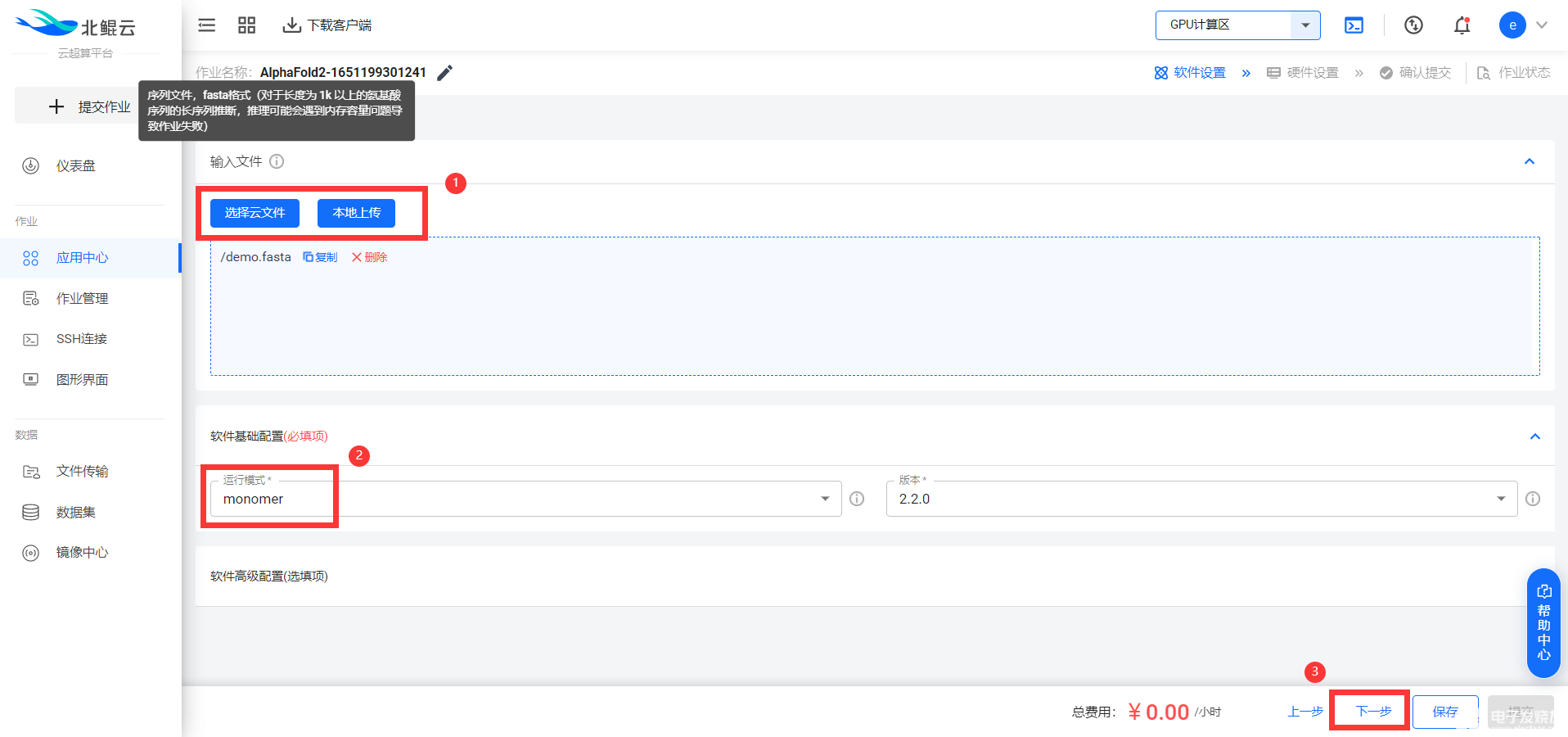
3. 填寫模板參數,選擇硬件配置,提交任務
上傳序列文件(.fasta格式),選擇運行模式(單體選擇monomer,多聚體選擇multimer)后即可點擊下一步:
選擇合適的GPU硬件配置后即可點擊下一步:
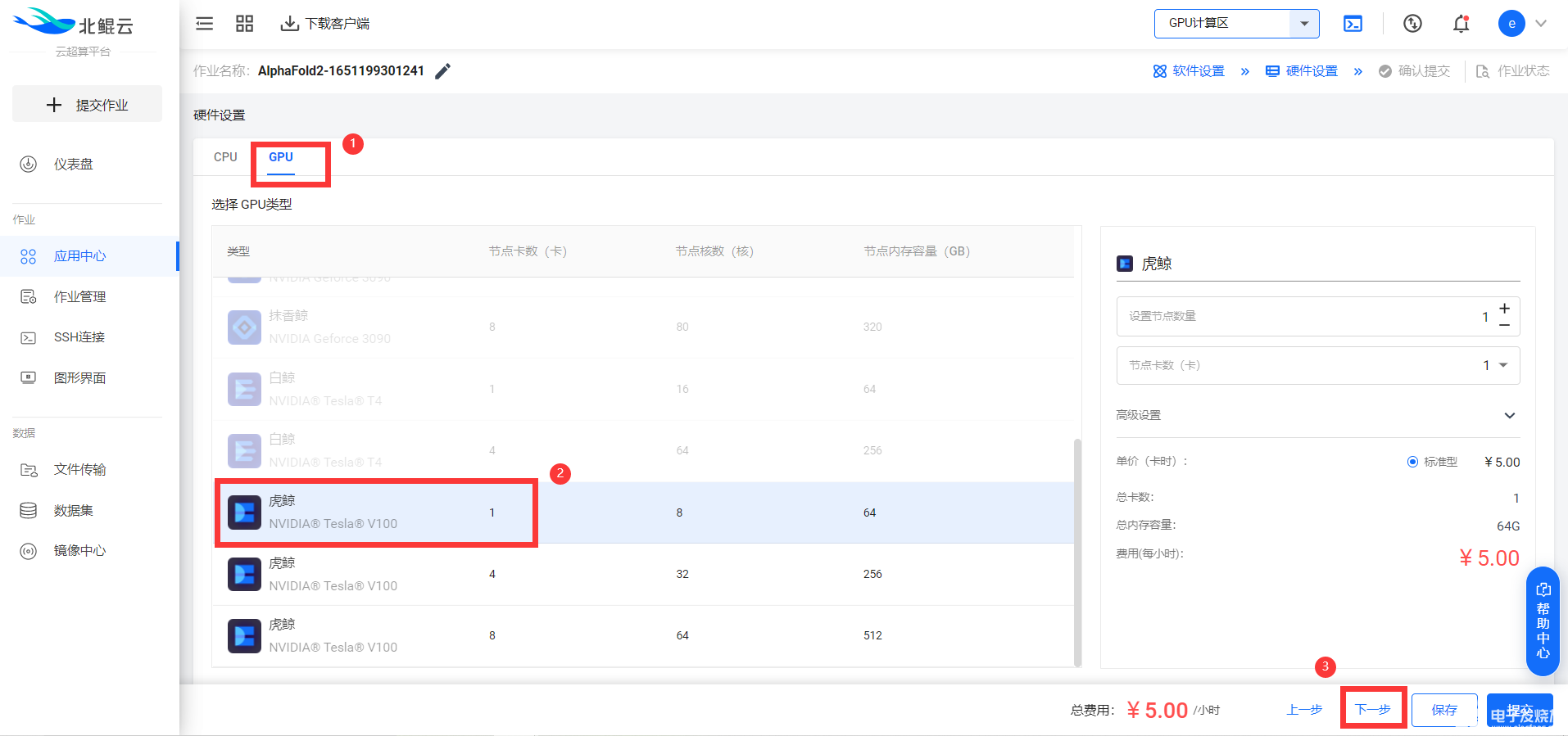
查看作業內容匯總并提交任務:
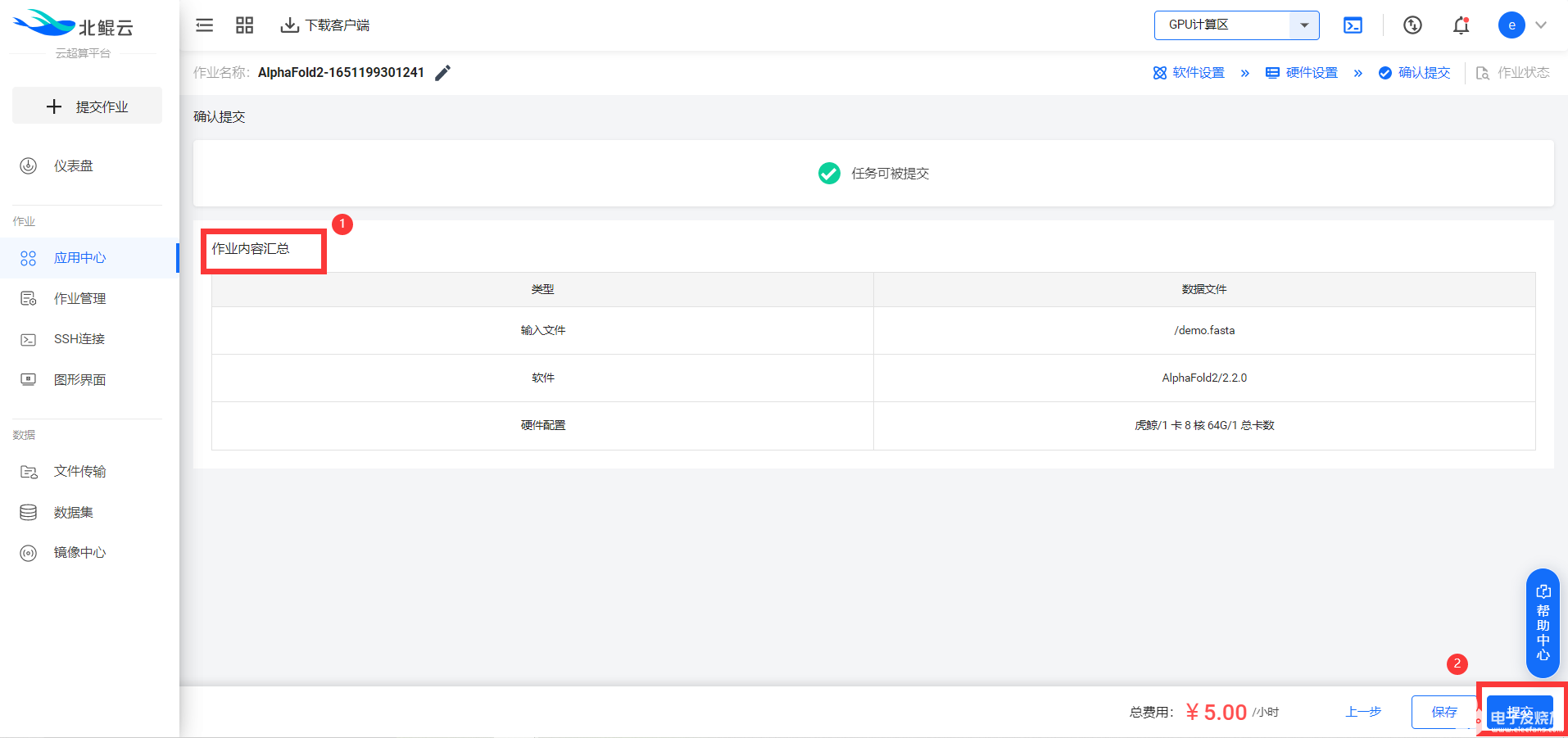
4. 查看任務詳情與結果
所有通過“模板”提交的作業,都可以在左側菜單欄“作業管理”功能中查看或者管理作業:
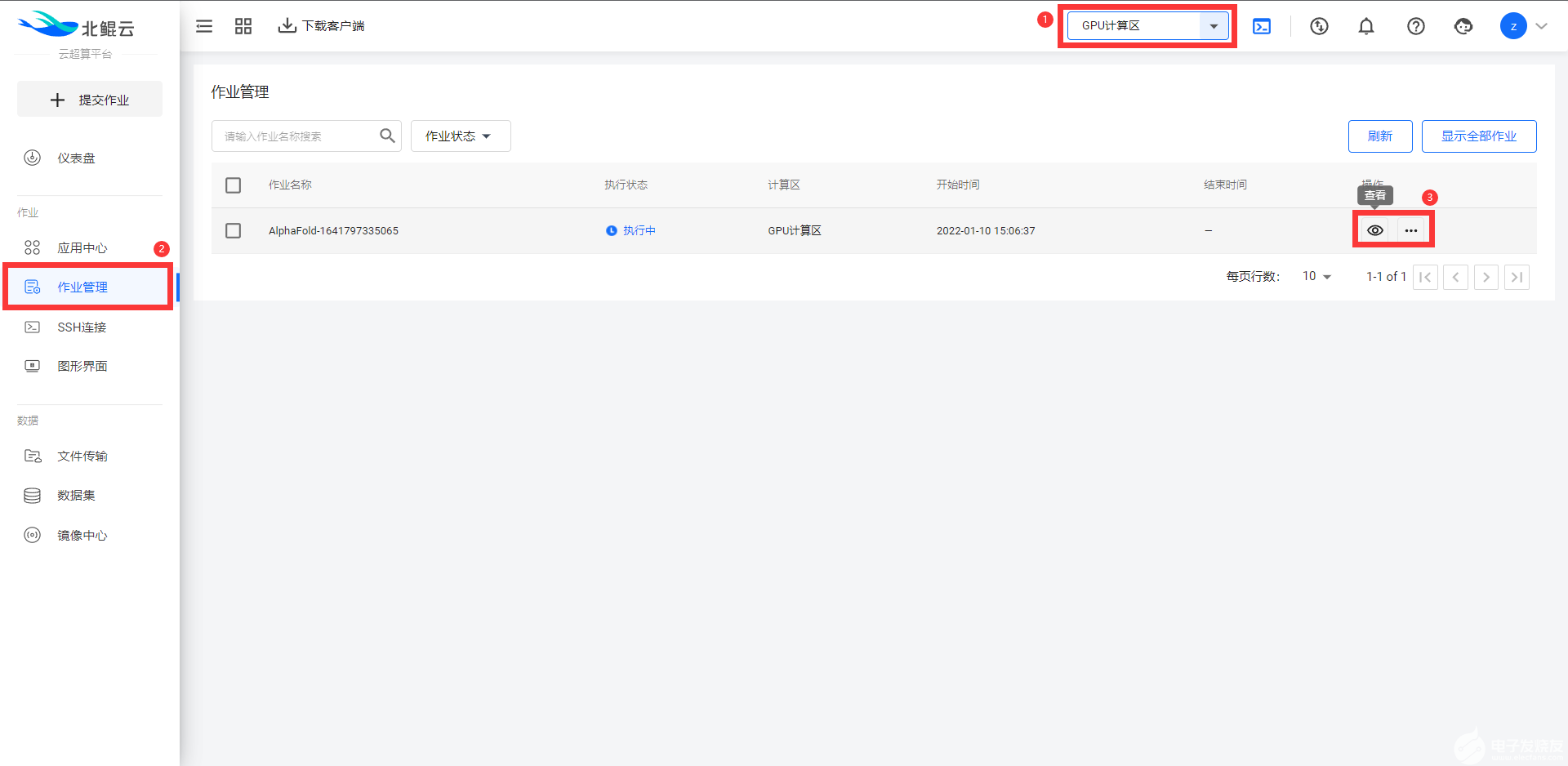
對于有Linux基礎和本地硬件配置足夠的人,本地使用AlphaFold2進行蛋白質預測的方法如下。
1. 配置要求
硬盤至少要3T以上,AlphaFold2訓練好的模型加數據庫下載下來是428 GB大小的文件,解壓后需要2.2T的空間。如果你用reduced_dbs(這個是簡化的數據庫),那么至少也得有600 GB的硬盤空間。
12個虛擬CPU
內存85GB及以上
1個Nvidia A100 或者Nvidia V100 GPU卡
2. 下載程序需要的數據庫、程序和模型
首先你得在github上面把這個AlphaFold2項目(https://github.com/deepmind/alphafold)給下載到一個本地目錄,然后進入scripts這個文件夾里面,運行命令download_all_data.sh <下載目錄>,程序會自動進行下載。
這個過程大概會下載438GB的文件,得等待很長時間,如果斷網的話,你還得把其它的都刪掉,重新下載。不建議直接運行這個主程序,可以利用多臺機器分個下載。當然你也可以使用下載工具提前下載好,然后再拷貝到服務器上面去解壓。
除了pdb_mmcif 這個文件之外,其它的都是可以提前下載。為什么這個文件不行?因為pdb網站并沒有提供壓縮的mmcif數據庫文件,每個都是小文件,必須得用同步的方式把pdb服務器上面的數據庫同步到本地才行,這一步建議直接在安裝目錄上去操作單獨腳本下載,不然到時候拷貝和壓縮以及解壓要花大力氣,這個文件夾里面有足足18萬個cif文件。
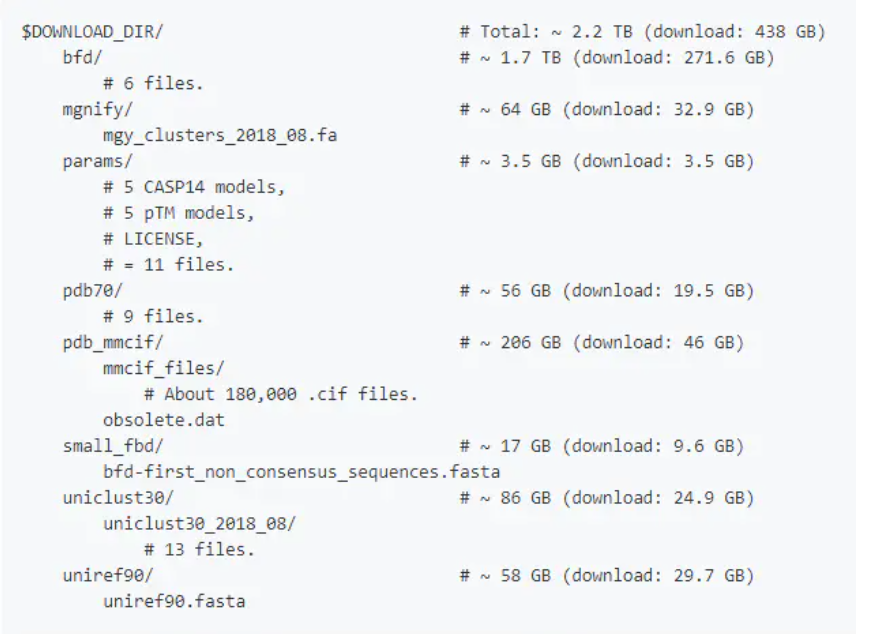
下載完成解壓后關注每個文件夾文件大小和文件名是否與上面這張圖中列出來的一致。
注意事項:bfd文件夾和small_bfd這兩個文件夾是互斥的,大文件夾里面只留一個,bfd是完整的數據庫而small_bfd是簡化的數據庫。如果你的磁盤不夠,你就下后者,271.6 GB的bfd文件你就別下了。
3. 安裝Docker和NVIDIA Container Toolkit
3.1 安裝Docker
參考Docker官方教程
3.2 安裝NVIDIA Container Toolkit
參考NVIDIA官方教程
3.3 測試是否安裝成功
root權限運行:
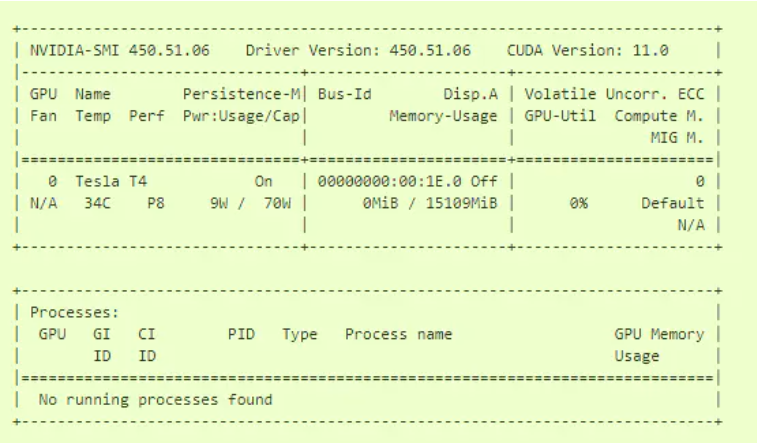
docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
如果你看到如下圖的一個表格,證明你成功了。
4. 使用AlphaFold2
4.1 配置輸入輸出文件夾路徑
首先你得配置一下輸入和輸出目錄,打開docker文件夾下的run_docker.py腳本,然后把其中的DOWNLOAD_DIR參數改成fasta文件夾的輸入目錄,把output_dir后面改為輸出結果的路徑。
4.2 docker build
docker build -f docker/Dockerfile -t alphafold
4.3 安裝pythin虛擬環境
如果你使用python3,并且機器里面有pip3,你可以直接:
pip3 install -r docker/requirements.txt
4.4 運行AlphaFold2
python3 docker/run_docker.py --fasta_paths=輸入序列文件完整路徑 --max_template_date=2020-05-14 --preset=[reduced_dbs、full_dbs、casp14]
fasta_paths:預測蛋白質fasta文件的文件名
max_template_date:如果你預測蛋白在pdb里面,而你不想用這個pdb做模板,你就用這個日期來限制使用該pdb做模板,這個日期應該早于這個蛋白結構的release date
preset:時間和預測質量的均衡考慮:reduced_dbs最快,但是質量最差,full_dbs中等,casp14質量最好但時間是full_dbs的八倍左右。
4.5 查看運行結果
運行結束后,在你的output_dir中會生成一系列文件,其中ranked_0到4就是AlphaFold2預測出來的分數最高的五個模型,0是最好的,可信度依次往下。
審核編輯 黃昊宇
-
人工智能
+關注
關注
1806文章
49000瀏覽量
249254 -
Alpha
+關注
關注
0文章
45瀏覽量
26204
發布評論請先 登錄






 工商網監
工商網監
評論